Desarrollo Embrionario y Anomalías Congénitas que Afectan a la Reproducción
Albert Fortuny
Unitat de Diagnóstic Prenatal. Hospital Clínic. Institut d´Investigacions Biomédiques August Pi Sunyer. Universitat de Barcelona. Hospital Clínic. Facultat de Medicina. Departament d´Obstetricia i Ginecologia. c/ Villarroel 170. 08036 Barcelona
INTRODUCCIÓN
Las anomalías congénitas que afectan la capacidad reproductora de la mujer responden a distintas causas que de algún modo afectan a elementos esenciales para completar el proceso reproductor. Aunque aisladamente no son frecuentes, en conjunto su impacto es importante y en algunos casos de difícil precisión en su origen.
El término congénito, en la práctica, se interpreta como presente en el nacimiento. Sin embargo no es infrecuente que la manifestación de determinadas anomalías no se haga evidente hasta la adolescencia, desarrollo puberal o edad adulta ante el intento reproductor.
Los términos congénito, genético y hereditario o transmible no deben considerarse sinónimos. Una anomalía congénita puede no ser necesariamente genética ni hereditaria sino por efecto ambiental esporádico o multifactorial por coincidencia de factores genéticos y ambientales.
En condiciones normales el proceso de formación del sistema reproductor se realiza a través de una secuencia bien programada y armónica de la determinación sexual, diferenciación de órganos y estructuras e integración de funciones. Cualquiera de estas etapas es susceptible de errores o interferencias que pueden conducir a incapacidad reproductora.
Existen, por otra parte errores metabólicos o enzimáticos congénitos que, sin afectar directamente a órganos y estructuras propiamente genitales, son capaces de interferir en el funcionalismo reproductor normal (Adrenal, Tiroideo, Metabolopatías, etc.).
DESARROLLO EMBRIONARIO NORMAL. DETERMINACIÓN Y DIFERENCIACIÓN SEXUAL
Determinación sexual
En la especie humana la determinación del sexo se establece en el momento de la fecundación. Los mecanismos que intervienen son esencialmente genéticos y vienen determinados por la distribución de los cromosomas X e Y en el embrión.
En el embrión 46,XX uno de los cromosomas X es precozmente inactivado en su mayor parte en la fase de implantación o quizás antes. Este cromosoma aparece como corpúsculo de Barr o cromatina sexual en las células somáticas en interfase. La inactivación no tiene lugar en las células germinales por lo que en los ovocitos ambos cromosomas permanecen activos.
La inactivación del cromosoma X depende de un gen localizado en el brazo largo del cromosoma X (Xq13), en la zona próxima al centrómero Xic (X inactivation center) y cuyo producto se denomina XIST (Avner and Heard, 2001). La inactivación de uno de los cromosomas X en cada célula es totalmente aleatoria, siempre que ambos cromosomas X sean normales, pudiendo afectar al cromosoma de origen paterno o materno pero, una vez inactivado, todas las células derivadas mantienen el mismo patrón de inactivación es decir, el Xpat. o el Xmat. Puesto que ello se produce cuando el embrión tiene unos centenares de células, las mujeres normales tienen dos poblaciones celulares, unas con Xpat. y otras con Xmat., tratándose por tanto de mosaicos con relación al cromosoma X activo. En algunos tejidos extra-embrionarios la inactivación no es aleatoria sino que es dictada por el origen paterno o materno, como es el caso de la placenta en la que el cromosoma X activo es siempre el de origen paterno.
En la especie humana, algunos de los loci del cromosoma X inactivado (heterocromático) permanecen sin embargo activos, particularmente en la parte distal del brazo corto (Xp) y brazo largo (Xq). Exceptuando los determinantes denominados de «mantenimiento ovárico», la mayoría de genes no inactivados tienen poco o nada que ver con el desarrollo reproductor.
Diferenciación gonadal
Las células germinales primordiales, aparecen en la 4ª semana de desarrollo en el endodermo del saco vitelino, migrando posteriormente a la cresta genital para inducir la formación de las gónadas indiferentes. No está bien establecido si las células somáticas de la gónada se originan en el epitelio celómico o en el mesonefros. En cualquier caso se produce una proliferación que da como resultado la formación de los cordones sexuales primitivos y de la gónada indiferente.
Las gónadas indiferentes se desarrollan como testículo si el embrión, o más probablemente el estroma gonadal, posee la dotación cromosómica XY. Este proceso se inicia alrededor de 43 días después de la concepción, y el testículo aparece morfológicamente identificable 7-8 semanas después de la concepción (9-10 semanas después de la última menstruación).
Bases genéticas de la diferenciación ovárica
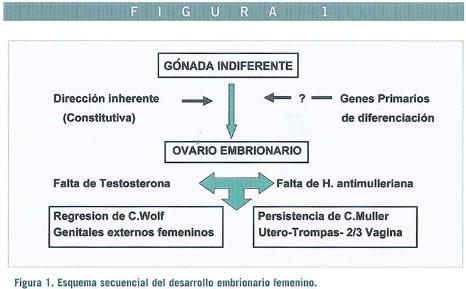
En ausencia de cromosoma Y la dirección inherente de diferenciación gonadal es en sentido ovárico. La presencia de un solo cromosoma X (monosomía X clínicamente traducida como síndrome de Turner) no parece interferir en el proceso de migración germinal en la vida fetal aunque la progresión ulterior normal del desarrollo ovárico exige la presencia de dos cromosomas X. La transformación en ovario se inicia entre los 50 y 55 días de desarrollo embrionario.
La dotación del ovario en células germinales (ovogonias) se expande por mitosis durante la vida fetal hasta el inicio del proceso de meiosis. Éste se interrumpe en la profase de meiosis I (ovocitos) rodeándose de una línea especializada de células del estroma (células pre-granulosa) para formar los folículos primordiales. Ello marca el fin de la producción de células germinales y los ovocitos no van a reanudar la meiosis hasta después de la ovulación y fertilización en la vida adulta.
La mayor parte de las células germinales producidas durante la ovogénesis mueren espontáneamente antes o poco después del nacimiento, de modo que de los 7 millones de células germinales del ovario fetal a las 20 semanas, permanecen sólo 1 o 2 millonesa de ovocitos viables en el período neonatal.
Este declive de la dotación germinal continua en la vida post-natal por un proceso continuo de atresia folicular de modo que en la pubertad la dotación queda reducida a alrededor de 300.000, y solo 400 de los ovocitos presentes en la pubertad sobreviven lo suficiente para completar el desarrollo y ser liberados para su potencial fecundación.
Este proceso de eliminación germinal se interpreta actualmente como un fenómeno de muerte celular programada (apoptosis) aunque la única evidencia es de carácter experimental y procede de la ratona en la que se ha identificado el gen Bax como inductor de lesión mitocondrial y mediador de la muerte celular de los ovocitos (Pru and Till, 2001O. En la monosomía X (45,X, síndrome de Turner) las células germinales se forman pero no pueden persistir y los folículos ováricos han degenerado ya en la vida postnatal. Por ello el segundo cromosoma X se considera importante para el mantenimiento pero no para la diferenciación ovárica.
Persiste la cuestión de si es suficiente para la diferenciación ovárica la ausencia de SRY (ver más adelante), es decir por defecto, o requiere un gen específico. Se ha propuesto el gen DAX1 localizado en Xp que, cuando está duplicado, puede re-dirigir embriones 46,XY hacia la diferenciación femenina, especulando si esta región DAX1 (Xp21) pudiese jugar un papel primario en la diferenciación ovárica. La evidencia actual, sin embargo, apunta a que la diferenciación ovárica es por defecto o pasiva, aunque deben existir «genes de mantenimiento» ovárico en distintas localizaciones, tanto en Xp como en Xq. (Figura 1).
Cromosoma Y en la diferenciación testicular
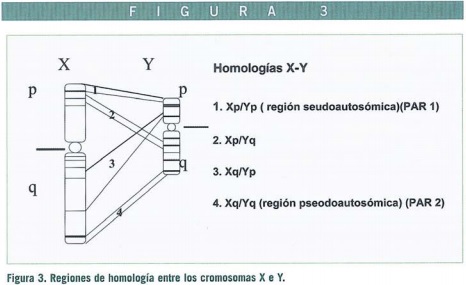
El cromosoma Y contiene regiones de DNA características. Los cromosomas sexuales humanos X e Y se originan hace algunos cientos de millones de años de un autosoma ancestral (no sexual) en el proceso evolutivo de la determinación sexual, divergiendo en las secuencias a lo largo de los milenios (Ohno, 1967). En el cromosoma Y actual existen regiones relativamente cortas a ambos extremos del cromosoma Y idénticas a las correspondientes en regiones del cromosoma X, reflejando el frecuente intercambio de DNA entre estas regiones (recombinación) que tiene lugar durante la espermatogénesis (Burgoyne, 1982).
Mientras las regiones distales de los cromosomas X e Y intercambian secuencias durante la producción de espermatozoides, el resto de secuencias de los cromosomas sexuales no lo hacen. Sin embargo, más del 95% del cromosoma Y (alrededor de 23 millones de pares de bases o Mb de eucromatina) es específico del sexo masculino, y una porción variable de heterocromatina (consistente en secuencias altamente repetitivas de DNA), son consideradas como no funcionales.
Recientemente, ha sido descifrada la secuencia del segmento eucromático del cromosoma Y, designando a este segmento como MSY (región masculino-específica de Y) (Skaletsky et al., 2003). El 10-15% de la región MSY consiste en secuencias que se trasladaron desde el cromosoma X en los últimos millones de años de evolución y son idénticas en el 99% a las secuencias presentes en el cromosoma X.
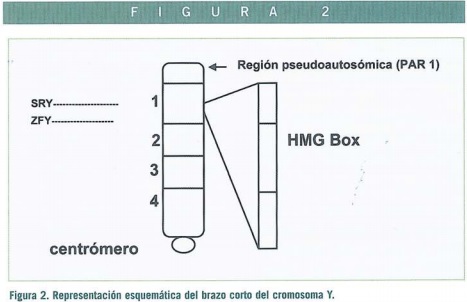
Hace más de dos décadas, se dedujo que el determinante genético esencial en la diferenciación testicular, actualmente denominado TDF (Factor testículo-determinante) se localizaba en el brazo corto del cromosoma Y (Yp). El gen clave es actualmente conocido como SRY (región testículo determinante de Y) (Figuras 2-4), se localiza en una de las regiones con homología X-Y. (Región Pseudo Autosómica o PAR).
Aproximadamente el 10-15% de casos esporádicos de disgenesia gonadal XY (hembras XY) muestran mutaciones del gen SRY. 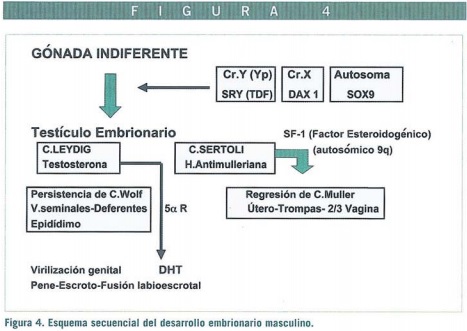 Sin embargo, la mayoría de hembras XY no muestran mutaciones en el gen SRY lo que sugiere que otros genes son necesarios para la determinación testicular. La función exacta del gen SRY al nivel celular en la diferenciación testicular no está claramente establecida en cuanto a su posible papel transcripcional o de interacción de productos del gen.
Sin embargo, la mayoría de hembras XY no muestran mutaciones en el gen SRY lo que sugiere que otros genes son necesarios para la determinación testicular. La función exacta del gen SRY al nivel celular en la diferenciación testicular no está claramente establecida en cuanto a su posible papel transcripcional o de interacción de productos del gen.
Existe actualmente cierta evidencia en experimentación animal (ratón) que sugiere una mayor complejidad del proceso, con intervención de genes como el DAX1 (en el cromosoma X y presumiblemente capaz de dirigir la diferenciación en sentido ovárico) y cuya duplicación podría frenar la acción del gen SRY en la inducción testicular (Meeks et al., 2003). Otras regiones autosómicas podrían ser importantes en la diferenciación testicular, aunque su relación con SRY es todavía menos clara, como es el caso del gen SOX9 en el cromosoma 17q, cuya delección es responsable de la disgenesia gonadal XY (reversión sexual) asociada a displasia campomélica y, en contraste, su duplicación da como resultado un individuo 46,XX con gónadas escrotales bilaterales, microfalo y orificio uretral perineal (un varón XX incompleto) (Huang et al., 1999).
Cromosoma Y y espermatogénesis
El brazo largo del cromosoma Y (Yq) en la región próxima al centrómero, contiene genes importantes para la espermatogénesis. Alrededor del 15% de varones azoospérmicos y 5-10% de oligospérmicos presentan pequeñas deleciones. El modelo actualmente más aceptado establece tres localizaciones de genes responsables de la espermatogénesis (genes AZF-a, b y c). La deleción de AZF-a es la menos común y se asocia a ausencia de espermatogonias. La deleción de AZF-b resulta en interrupción de maduración espermática. El gen AZF-c contiene el locus DAZ (deleted in azoospermia) y su deleción produce azoospermia u oligospermia, aunque los varones con oligospermia severa pueden actualmente conseguir gestaciones mediante inyección intracitoplásmica de espermatozoides (ICSI).
Diferenciación de conductos genitales
Las estructuras embriológicas primordiales durante el estadio indiferente consisten en dos pares de conductos, mesonéfricos (conductos de Wolf) y paramesonéfricos (conductos de Müller) que coexisten en estado indiferente durante las primeras semanas (Skaletsky et al., 2003; Meeks et al., 2003; Huang et al., 1999; ASRM, 1988).
Los conductos de Müller se originan como invariaciones longitudinales del epitelio celómico en la cresta genital cuyas partes caudales contactan y se fusionan para formar el primordio uterovaginal y contactan a su vez con la pared posterior del seno urogenital. En ausencia de factor inhibidor mulleriano (MIF o AMH), producido por las células de Sertoli, los conductos forman el útero, trompas y probablemente la parte superior de la vagina.
Los genitales externos inician su desarrollo durante la 4ª semana a partir de células del mesénquima que migran hacia la membrana cloacal formando los pliegues denominados eminencias labioescrotales.
Mientras el efecto gonadal en los conductos es de tipo local, como se demostró en los clásicos experimentos de Jost, el efecto en los genitales externos depende de las substancias hormonales que alcancen estas estructuras y de la presencia de receptores específicos. En el embrión hembra la ausencia de testosterona y MIF permite que los conductos de Wolff regresen, y no se masculinizen los genitales externos indiferentes, dando lugar a los labios mayores y menores, y el clítoris a partir del tubérculo genital.
El extremo caudal o distal de los conductos de Müller fusionados contacta con el seno urogenital causando proliferación de los bulbos del seno endodérmico que, a través de una evaginación formará la placa vaginal. Esta proliferación continua en ambos sentidos, craneal y caudal, aumentando la distancia entre el útero en desarrollo y el seno urogenital (semana 13) y alrededor de la semana 17-18 se forma una cavidad entre el tubérculo del seno urogenital y los conductos de Müller fusionados. Se acepta que a los 5 meses de gestación la vagina está completamente canalizada, siendo su parte alta (fornix) derivada de los conductos de Müller y la parte baja del seno urogenital. El himen separa el seno urogenital de la vagina y consiste en una capa de células vaginales y una capa de células epiteliales del seno urogenital (Tablas I-II).
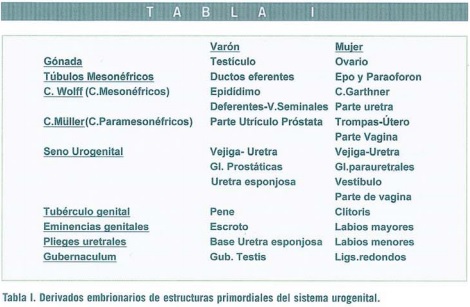

Los conductos de Müller crecen paralela y externamente a los conductos de Wolff hasta cruzarlos anteriormente y encontrarse en la línea media para fusionarse y eventualmente canalizarse. La parte más cefálica de los conductos de Müller da lugar a las trompas. La íntima relación entre los conductos de Müller y Wolff (mesonefros y conductos mesonéfricos) explica la frecuente asociación de anomalías mullerianas y renales (mesonéfricas), estimada entre 20-30%, particularmente la asociación de útero unicorne y agenesia renal unilateral. Por ello se recomienda la evaluación urológica sistemática.
Anomalías en el desarrollo del sistema reproductor femenino
La mayoría de anomalías observadas clínicamente responden a la interrupción en el desarrollo de las estructuras primordiales mullerianas uterovaginales alrededor de la 8ª semana de desarrollo, ya sean defectos de fusión o migración que generan anomalías estructurales de trompa, útero y 2/3 superiores de la vagina (Tabla III).
Existen, sin embargo, excepciones en las que no se trata simplemente de interrupción en una fase normal del desarrollo como es el caso de la presencia de dos úteros y dos cuellos uterinos.
La prevalencia global de anomalías estructurales mullerianas se estima entre 2 y 3%, y su incidencia real en mujeres en edad reproductora es difícil de estimar, dado que su identificación depende de los métodos de diagnóstico utilizados y sólo el 25% plantean problemas de fertilidad. La aplicación reciente de técnicas de imagen con mayor poder de definición ha permitido aumentar el diagnóstico en anomalías que de otro modo pasarían desapercibidas al examen clínico.
La etiología de la mayoría de estas anomalías es desconocida aunque por su baja frecuencia individual y carácter esporádico, sugieren mecanismos poligénicos o multifactoriales, y en algunos casos se ha demostrado un factor ambiental como el efecto del dietilestilbestrol o la thalidomida. La mayoría de malformaciones uterinas congénitas muestran, sin embargo, un cariotipo 46, XX.
Existen distintas anomalías del sistema ductal femenino (Tabla III) cuya clasificaciòn ha sido confusa, aceptándose actualmente la revisada por la ASRM (ASRM, 1988) (Tabla IV).


Las anomalías aisladas del desarrollo embrionario del cuello uterino son raras. La agenesia y atresia cervical aisladas se interpretan como fallos segmentarios de canalización del primordio uterovaginal. Lo mismo ocurre con las anomalías de las trompas cuya agenesia parcial o total, uni o bilateral, son de rara observación (Lin et al., 2002).
La ausencia congénita de vagina y otras estructuras mullerianas, con cariotipo, gónada y caracteres sexuales normales, a veces con forma de duplicidad uterina, son características del síndrome de Mayer-Rokitansky-Kuster-Hauser.
La atresia parcial de vagina y tabiques o septos vaginales transversos son defectos parciales de canalización, o fallo de los conductos de Müller fusionados en contactar con el seno urogenital. La falta de unión del primordio vaginal (endodérmico) con el conducto vaginal (mesodérmico) puede ocurrir a cualquier nivel del canal vaginal, aunque es más frecuente en el tercio medio y superior. En contraste, son el resultado de la fusión caudal incompleta de los conductos de Müller y pueden ser comunicantes u obstruidos.
El himen imperforado no es de origen mulleriano sino que deriva del tubérculo del seno urogenital y representa el límite entre ambas estructuras embrionarias.
Disgenesias gonadales
El concepto genérico de disgenesia gonadal incluye distintas formas que tienen en común el desarrollo gonadal anómalo. Se trata, por tanto, de anomalías en la diferenciación gonadal.
Su clasificación es, sin embargo, compleja dado que, con frecuencia, se asocian factores genéticos y consecuencias hormonales que conducen a manifestaciones fenotípicas de ambigüedad genital diversas y por ello se las incluye en el concepto de estado intersexual o intersexo. Es por ello, importante destacar que disgenesia gonadal e intersexo son conceptos distintos y serán por tanto tratados separadamente, aunque en ocasiones pueden coincidir en un determinado individuo.
La Academia Internacional de Patología establece una clasificación útil ya que introduce los epígrafes de dotación cromosómica sexual que incluye los seudohermafroditismos, y en la dotación cromosómica sexual anormal distingue la ausencia o presencia de ambigüedad sexual (Tabla V).

DISGENESIA GONADAL Y FALLO OVÁRICO PRIMARIO
Monosomía X
Actualmente el término «disgenesia gonadal» se aplica convencionalmente a las mujeres que presentan rudimentos gonadales descritos como «cintillas ováricas» (streak gonad). El epónimo síndrome de Turner se aplica a los casos que muestran estatura baja asociada a otras anomalías somáticas (estigmas Turner). La presencia de estigmas Turner no siempre implican la presencia de cintilla gonadal, por lo que el síndrome de Turner se limita a individuos con disgenesia gonadal, estigmas Turner y cariotipo 45,X (monosomía) o deleciones parciales del cromosoma X, es decir monosomías parciales.
En el 80% de casos 45,X el cromosoma X es de origen materno (Xm) y el 20% de origen paterno (Xp). La disgenesia gonadal en la monosomía X no es un defecto en la formación de células germinales, que de hecho existen normalmente en el feto hembra hasta prácticamente la mitad de la gestación, sino de un proceso acelerado de atresia folicular, y por tanto un defecto de mantenimiento ovárico que conduce, en la vida postnatal, a la desdotación germinal con sólo la presencia de tejido fibroso análogo al estroma ovárico.
El patrón hormonal es, en consecuencia, de hipogonadismo hipergonadotrófico, y las manifestaciones en los caracteres sexuales secundarias derivadas del déficit de esteroides ováricos.
Aunque se han descrito algunos casos de 45,X que inician menstruación espontánea, lo más plausible es la existencia de líneas celulares 46,XX no detectadas en el estudio citogenético.
La terapia de la infertilidad en pacientes 45,X conlleva soporte hormonal para crear las condiciones uterinas esenciales, fecundación asistida con donantes de óvulos y transferencia embrionaria al útero hormonalmente sincronizado. 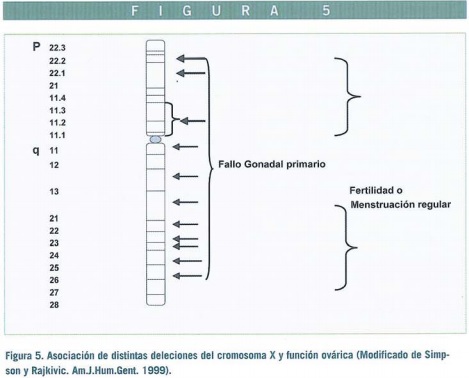 El éxito en obtener una gestación clínica es de alrededor del 20% aunque el índice de abortos espontáneos es elevado (más del 50%) (Foudila et al., 1999). Este elevado índice de aborto refleja probablemente más causas hormonales o uterinas que la transmisión de gametos aneuploides.
El éxito en obtener una gestación clínica es de alrededor del 20% aunque el índice de abortos espontáneos es elevado (más del 50%) (Foudila et al., 1999). Este elevado índice de aborto refleja probablemente más causas hormonales o uterinas que la transmisión de gametos aneuploides.
La región o regiones del cromosoma X responsables de evitar los estigmas Turner no están bien esclarecidas, pero no necesariamente coinciden con la región de mantenimiento ovárico, y la parte distal de Xp ha sido particularmente implicada (Figura 5). Los genes candidatos en Xp o Xq que podrían ser importantes para las repercusiones ováricas y somáticas han sido consideradas por Bione y Toniolo (2000). Los fetos 45,X tiene bajo peso al nacer, el índice de crecimiento antes de la pubertad se sitúa alrededor del percentil 10-15 y la talla media adulta oscila entre 140-160 cm. Se han propuesto diversos tratamientos para la baja estatura (esteroides anabólicos, hormona de crecimiento, estrógenos a dosis baja) todos ellos con beneficio observable particularmente poco después de iniciar el tratamiento y, aunque el resultado final es menos ostensible, se consiguen incrementos entre 6-8cm (Rosenfeld and Grumbach, 1990). Los resultados más favorables parecen obtenerse con la hormona de crecimiento derivada de DNA recombinante humano (Rosenfeld et al., 1998).
Mosaicismos
El concepto de mosaicismo o mosaico implica la presencia en un mismo individuo de más de una línea celular procedente del mismo cigoto. Ello se produce por errores en la división, ya sea por no-disyunción o por retraso en anafase, y si ocurre en el cigoto o en el embrión puede aparecer más de una línea celular. El complemento cromosómico final depende del estadio de desarrollo en que se produjo el error y de la capacidad de supervivencia de las líneas celulares resultantes.
La habilidad en reconocer la presencia de mosaicos depende del número de células analizadas por tejido y del número de tejidos analizados. La práctica común de analizar 20 células por tejido conlleva una probabilidad del 12% de no detectar por lo menos una célula de una línea representada en el 10% de la población celular. La eficacia en detección aumenta con el porcentaje de la línea y con el número de células examinadas.
Los mosaicos más comúnmente observados asociados a la disgenesia gonadal son los 45,X/46,XX. Sin embargo, en estos casos, no es infrecuente que inicien menstruación espontánea y eventualmente fallo ovárico precoz indicando una dotación folicular reducida y/o atresia acelerada. Menos común, pero fenotípicamente similar al mosaico 45,X/46,XX es el 45,X/47,XXX.
Cuando el mosaico incluye una línea celular 46,XX también puede mostrar cintilla gonadal bilateral, pero lo más común es la presencia de cintilla gonadal unilateral y testículo disgenético, recibiendo en tal caso el nombre de disgenesia gonadal mixta que, por presentar ambigüedad genital, se incluye como una forma de seudohermafroditismo.
Deleciones del brazo corto del cromosoma X y fallo gonadal
Estos casos muestran un fenotipo variable en función del segmento de brazo corto que permanece. El punto de rotura más frecuente es Xp 11.2-11.4 (Figura 5) permaneciendo sólo la parte próxima del brazo corto cerca del centrómero. La mitad de los casos 46,X, del (Xp) (p11) muestran amenorrea primaria y disgenesia gonadal, y el resto amenorrea secundaria.
Las deleciones terminales (p22.1 ó 22.2) todavía ven afectado el desarrollo ovárico, pero la deleción de la parte más distal (p22.3) no muestran amenorrea. Aunque se están llevando a cabo estudios moleculares para mayor precisión, hasta el momento la única conclusión es que la amplia zona Xp11.3-22.1 es, probablemente, la más importante para el desarrollo ovárico.
Isocromosoma X
Cuando la división del centrómero no es longitudinal sino transversal da lugar a un isocromosoma con duplicación de brazos largos [i(Xq)] o cortos [i(Xp)], aunque esta última forma se considera inexistente. El [i(Xq)] es la anomalía cromosómica estructural más común, aunque con frecuencia coexisten en mosaico con céluluas 45,X. Presentan disgenesia gonadal y con frecuencia estigmas Turner.
Deleciones del brazo largo del cromosoma X
La región de «mantenimiento ovárico», mencionadas previamente, se sitúa en Xq13 y no más allá de q21 (Figura 5). Las deleciones afectan zonas más distales no se presentan con amenorrea primaria sino por fallo ovárico prematuro, por lo que esta zona parece menos importante para el mantenimiento ovárico, aunque si pueda tener cierta participación sin que se haya todavía establecido una clara demarcación.
Polisomías del cromosoma X y fallo gonadal
La presencia de más de un cromosoma X en la mujer es relativamente frecuente (1:800-1000 nacidos vivos). Aunque invariablemente causan anomalías gonadales en individuos con cromosoma Y (47,XXY o síndrome de Klinefelter), sólo ocasionalmente lo hacen en mujeres 47,XXX (triple X) o en varones 47,XYY.
En la mujer 47,XXX el origen es comúnmente en la meiosis I materna, el fenotipo es normal y en la mayoría sin anomalías somáticas. Como grupo, existe mayor riesgo de retraso mental discreto y sólo el 5% muestran C.I. entre 45-70. La función gonadal puede verse afectada en forma de menarquia tardía y/o fallo gonadal prematuro (menopausia precoz). Se desconoce el origen de la disfunción ovárica pero se presupone que el cromosoma X adicional interfiere en la segregación meiótica conduciendo secundariamente al cese de la ovogénesis, de igual modo como ocurre en el caso 45,X o en las trisomías autosómicas 13 y 18.
La mayoría de descendientes de mujeres 47,XXX son normales aunque se han observado algunas anomalías (Simpson, 1981). En la tetrasomia X (48,XXXX) se observa retraso mental manifiesto y disfunción ovárica. En las escasas observaciones de pentasomía X (49,XXXXX) la presencia de anomalías somáticas, particularmente faciales y extremidades, así como el retraso mental son prácticamente constantes.
Fallo ovárico de causa autosómica
Existen diferentes genes autosómicos capaces de producir fallo ovárico entre las que cabe citar:
1. Disgenesia gonadal 46,XX no asociada a alteraciones somáticas excepto por la afectación gonadal. Se considera que 2/3 de estas disgenesias tienen causa génica de tipo autosómico recesivo aunque los genes no han sido identificados. El resto, denominadas fenocopias, pueden ser debidas a causa ambiental (infección, necrosis, infiltración autoinmune).
2. Disgenesia gonadal XX asociada a sordera neurosensorial (síndrome de Perrault) transmitida en forma de autosómica recesiva.
3. Disgenesia gonadal XX debida a mutación de receptor FSH, detectada predominantemente en Finlandia. En el brazo corto del cromosoma 2 (2p) se encuentran genes para los receptores de FSH y LH.
4. Mutación en el gen del receptor LH. Localizado en el cromosoma 2 (2p) puede causar oligomenorrea o con menor frecuencia amenorrea primaria. La gametogénesis prosigue hasta estadios pre-ovulatorios pero no existe ovulación.
5. Disgenesia gonadal XX y ataxia cerebelar es una forma heterogénea en la que es improbable que esté involucrado un gen único.
6. Disgenesias gonadales XX asociadas a otros síndromes malformativos han sido descritas (Tabla VI).

Disgenesia gonadal en mujeres 46,XY
En individuos con cariotipo masculino aparentemente normal la pérdida o ausencia de tejido testicular antes de las semanas 7-8 produce el fenotipo previsible. El prototipo de mujer XY con disgenesia gonadal muestra genitales externos femeninos normales, vagina, útero y trompas, y en la pubertad no hay desarrollo de caracteres sexuales secundarios, la estatura es normal y no se observa anomalías somáticas, por lo que el aspecto externo no difiere de la disgenesia gonadal XX.
El riesgo de desarrollo tumoral antes de los 20 años (Disgerminoma, Gonadoblastoma) es elevado (20-30%) por lo que se recomienda la extirpación gonadal y la gestación es posible mediante reproducción asistida con donante de ovocitos o de embriones (Kahn et al., 1997). Existen una serie de situaciones, descritas como «reversión o inversión sexual», es decir mujeres XY asociadas a distintos síndromes. (Tabla VII).
En las formas que no presentan anomalías somáticas, la etiología puede residir en la mutación o deleción del gen SRY y ello se constata en alrededor del 15% (figura 1). El resto corrrespondería a algunas de las formas descrita en la tabla y a las de causa no determinada.
En la hipoplasia de células de Leydig hay un insensibilidad completa a la LH (mutación del receptor LH) observándose testículos bilaterales desprovistos de células de Leydig, los genitales externos son femeninos pero no existe útero (por efecto del MIF, hormona antimulleriana que es operativa), sino deferentes y epidídimo. El gen de R-LH se localiza en el cromosoma 2 (Sultan and Lumbroso, 1998). Existen formas de insensibilidad o resistencia parcial que conducen a varones con pene hipoplásico o hipospadias.
En el déficit del factor esteroidogénico (SF-1) el gen (FTZ1) se localiza en el brazo largo del cromosoma 9 (9q33) (Figura 4) (Achermann, 1999). Muestran fallos suprarrenal, cariotipo 46,XY, cintillas gonadales, derivados mullerianos normales y genitales externos femeninos.
Disgenesia gonadal y trisomías autosómicas
Excepto la trisomía 21, los nacidos con trisomías se caracterizan por disgenesia ovárica. En las trisomías más comunes (cromosoma 18 y cromosoma 13) raramente se observan ovocitos, incluso a abortos de segundo trimestre. Ello se ha atribuido a alteraciones inespecíficas de la meiosis, aunque no se descarta que los cromosomas 13 y 18 puedan contener genes específicos para el control de la ovogénesis
Agonadismo
La ausencia de gónada ocurre generalmente en individuos 46, XY, no existiendo ni siquiera rudimentos (cintillas) de la gónada. Los genitales externos son anormales pero de tipo femenino, con falo del tamaño de un clítoris, hipoplasia de labios mayores y fusión labio-escrotal y presencia sólo de rudimentos mullerianos o wolfianos. Por su presentación, entraría en el concepto de seudohermafroditismo masculino pero la ausencia de gónada no permite incluirlo en la clasificación clásica (ver estados intersexuales). Se presupone, la presencia transitoria de testículo (vanishing testis), suficiente para frenar el desarrollo mulleriano pero no para completar la diferenciación masculina.
Hipogonadismos hipogonadotróficos
Los defectos congénitos que impiden la producción de FSH y LH conducen a la falta de esteroides sexuales y gametogénesis incompleta, desarrollo pubertal inadecuado, amenorrea primaria e infertilidad. La etiología es diversa reflejando alteraciones hipotalámicas (GnRH), hipofisarias (FSH-LH) o ambas.
Son formas de trasmisión mendeliana recesiva, ya sea ligada al cromosoma X (síndrome de Kallmann) o formas autosómicas (déficit aislados de FSH o LH) (Tabla VIII).
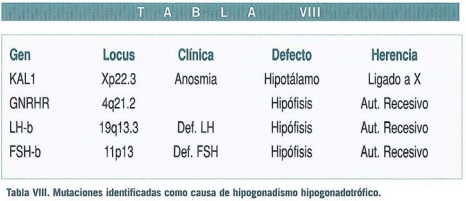
La forma más frecuente de hipogonadismo hipogonadotrófico es el síndrome de Kallmann debida al gen KAL1 (en la parte distal del cromosoma X que no sufre inactivación) y produce una proteína (Anosmina) que forma el entramado por el que migran las neuronas GnRH y los nervios olfatorios hacia el hipotálamo durante la embriogénesis. El fallo de esta proteína impide la migración neuronal dando como resultado el defecto hipotalámico asociado a la anosmia.
Otras formas de causa hipotalámica puede estar asociadas a anormalidades en la leptina o en sus receptores, asociándose comúnmente a obesidad (Montague et al., 1997). Los defectos aislados de FSH son menos comunes y pueden afectar tanto a varones como mujeres (Lyaman, 1999) pero los defectos aislados de LH sólo han sido descritos en el varón.
Estados intersexuales
El concepto clásico de «estado intersexual» lo define como la presencia en un mismo individuo de caracteres sexuales de ambos sexos, considerando dos tipos básicos, el seudohermafrodismo (o hermafroditismo verdadero) y los seudohermafroditismos.
Hermafroditismo verdadero
Viene definido por la coexistencia en un mismo individuo de tejido testicular y ovárico en cualquiera de sus variantes, uni o bilateral (testículo, ovárico, u ovotestes), y su localización puede ser pélvica, inguinal o labioescrotal, aunque en testículo u ovotestes sólo raramente se observa espermatogénesis.
El complemento cromosómico sexual es 46,XX en el 60% de casos descritos y en el resto distintos mosaicismos, siempre incluyendo de alguna forma una línea con cromosoma Y.
La forma 46,XY/46,XX es comúnmente atribuible a quimerismo que, a diferencia del mosaicismo, es la presencia en un mismo individuo de dos o más líneas celulares derivadas de distintos cigotos.
En los casos 46,XX la explicación de la presencia de testículo en individuos que carecen de cromosoma Y incluye la translocación durante la meiosis paterna del gen SRY del cromosoma Y al cromosoma X, translocación de SRY a un autosoma, mosaicismo no detectado o genes autosómicos implicados en la reversión sexual que explicarían las formas de transmisión mendeliana (formas familiares) de hermafroditismo verdadero 46,XY.
Los genitales externos muestran diversos grados de ambigüedad o predominantemente femeninos. Se observa la presencia de útero aunque frecuentemente uni o bicorne, y la ausencia de un cuerno uterino indica la vecindad homolateral de testículo u ovotestes.
Se han descrito una docena de casos de gestación en casos 46,XX y sólo un caso en mosaico 46,XX/46,XY (Verp et al., 1992).
Pseudohermafroditismos
Estos casos incluyen individuos con concordancia en el complemento cromosómico sexual y la gónada, pero que presentan defectos de masculinización en el caso del varón, o virilización en la mujer. No se trata, por tanto, de alteración en la diferencia gonadal, como en el caso del hermafroditismo verdadero, sino alteraciones manifiestas en los genitales externos y caracteres sexuales secundarios (Tabla V).
A. Seudohermafroditismo femenino
Definido por individuos 46,XX cuyas gónadas son ovarios y los genitales externos muestran cierta ambigüedad por masculinización.
La forma más común es la hiperplasia adrenal congénita como resultado del déficit de distintos enzimas necesarios para la esteroidogénesis (Figura 6): 21-hidroxilasa, 11B-hidroxilasa y 3Bhidro-esteroideshidrogenasa (3B-HSD) y aromatasa. Estos defectos se transmiten con carácter autosómico recesivo y los genes se han localizado respectivamente en el cromosoma 6 (CYP21) (6p21), en el cromosoma 8 (CYP11B1) (8q22) y en el cromosoma 1. El diagnóstico molecular es posible incluyendo el diagnóstico prenatal mediante biopsia corial y la terapéutica materna precoz con corticoides cuyo paso transplacentario es capaz de evitar o minimizar el efecto virilizante.
La patogenia común es el déficit de cortisol cuyo efecto de retro-alimentación (feed-back) negativo regula la producción de ACTH. La falta de inhibición en la secreción de ACTH conduce al aumento de los precursores esteroideos a partir de los cuales se sintetizan andrógenos cuyo aumento produce la virilización de fetos hembras.
El déficit de cortisol y corticosterona puede resultar en pérdida de sal con afectación severa al nacer en el 75% de casos de déficit de 21-hidroxilasa con virilización. En los casos sin pérdida de sal se asume que el incremento de ACTH produce suficiente cortisol y aldosterona para evitar esta complicación.
La forma más común es el déficit de 21-hidroxilasa que convierte la 17x-hidroxiprogesterona (17x-OHP) a 11-deoxicortisol (DOC) con el incremento resultante de precursores (17x-OHP, androstendiona, estrona y testosterona). Existen formas de inicio tardío con descenso enzimático parcial (entre 20-50%) que se manifiestan por exceso androgénico, anovulación y ovarios de tipo poliquístico. El déficit de 11B-hidroxilasa impide el paso de 11-deoxicorticosterona a corticosterona y de 11-deoxicortisol a cortisol.
En el feto hembra ambas formas se manifiestan por hipertrofia de clítoris, fusión labioescrotal y desplazamiento del orificio uretral, aunque el grado de virilización puede variar entre individuos con el mismo déficit enzimático. Las estructuras mullerianas se desarrollan normalmente dado que no existe factor antimulleriano testicular. Estos déficit en varones no modifican los genitales externos, pero a partir de los 2 años pueden mostrar aumento genital, vello púbico y aumento prematuro de estatura (pubertad prematura isosexual).
En el déficit de 3B-HSD el principal andrógeno producido es la dehidroepiandrosterona (DHEA), de acción androgénica débil, que no puede transformarse a androstenediona o testosterona por lo que la virilización de fetos hembra es mucho menor. Cuando afecta al feto varón no se masculiniza completamente y se presenta como un seudohermafroditismo masculino. La forma de déficit completo resulta en severa pérdida de sal.
Por último, el déficit de aromatasas esencial para la conversión de andrógenos _^4_androstenediona) a estrógenos (estrona), conduce a un exceso de androstendiona capaz de virilizar al feto hembra. El gen ha sido identificado (CYP19).
Existen formas teratógenas de seudohermafroditismo femenino, aunque para interferir en la diferenciación genital debe actuar durante la organogénesis. El tubérculo genital se hace evidente alrededor de las 5 semanas post-fecundación (semana 7 de gestación) y si se administran teratógenos androgénicos antes de la semana 12 de gestación pueden producir fusión labioescrotal, hipertrofia del falo o desplazamientos en la invaginación urogenital. A partir de las 12 semanas la única manifestación androgénica genital es la hipertrofia de clítoris.
Los teratógenos androgénicos incluyen la testosterona, etiniltestosterona y danazol. La nor-etindrona (19-nortestosterona) sólo se manifiesta a dosis elevadas de 10-20 mg/día por lo cual las dosis utilizadas en contraceptivos orales no conllevan riesgo de seudohermafroditismo.
Algunos tumores virilizantes en la gestante (Arrenoblastoma, Tumor de células de Leydig, luteomas) pueden virilizar el feto hembra, aunque son formas extremadamente raras coincidiendo con la gestación porque tales tumores interfieren con la fertilidad
B. Pseudohermafroditismo masculino
Clásicamente se definen como individuos con cariotipo 46,XY, presencia de testículos y manifestaciones de ambigüedad genital por deficiente masculinización.
Como una excepción a la del cariotipo 46,XY se consideran los casos de mosaico 45,X/46XY que varían en su expresividad fenotípica, desde varones casi normales con criptorquidia o hipospadias a ambigüedad genital o fenotipo femenino normal. Se asume que la línea 45,X es el resultado de la pérdida de un cromosoma Y estructuralmente anormal que se piede por su inestabilidad (Simpson, 1976; McDonough and Tho, 1983; Rosenberg et al., 1987). Se han descrito distintos tipos de alteraciones como causa de seudohermafroditismo masculino (Tabla V).
Déficits en la síntesis de Testosterona
La patogenia común es la de niveles insuficientes de testosterona para virilizar los genitales externos. El déficit enzimático involucrado puede ser cualquiera de los necesarios para llegar a la síntesis androgénica (A,B,C,E) (Figura 6) y se transmite con carácter autosómico recesivo.
a. Hiperplasia adrenal de células lipoides
No es posible la conversión de colesterol a pregnenolona. Muestran genitales ambiguos o de tipo femenino, marcada pérdida salina y las suprarrenales se caracterizan por la presencia de células espumosas llenas de colesterol. En humanos, es el resultado de la alteración del gen que codifica una proteína (STAR o steroidogenic acute regulatory protein) que conduce a los precursores de colesterol para el clivaje de su cadena lateral). Este gen se localiza en el cromosoma 8 (8p11.2).
b. Déficit de 3b-HSD
La síntesis de andrógenos y estrógenos está reducida produciéndose sólo DHEA con débil andrógenica e incapaz de virilizar adecuadamente el feto varón y además se presenta pérdida de sal por déficit de cortisol y aldosterona. Se han identificado 5 genes relacionados de los que sólo el tipo II se expresa en la suprarrenal y la gónada
c. Déficti de 17a-OH-asa y 17,20-desmolasa
Funciones asumidas por un único enzima, los afectados muestran genitales ambiguos o de tipo femenino, pero los derivados wolfianos y el desarrollo testicular son normales.
d. Déficit de 17b-HSD (17-cetosteroide Reductasa)
Conduce a la incapacidad de convertir DHEA a testosterona y los afectados muestran un desarrollo genital similar al anterior, pero en la pubertad muestran una mayor virilización que en los casos anteriores por lo que el sexo social comúnmente se cambia a varón después de la pubertad (Morris and Mahesh, 1963). Esta mayor virilización puberal, que intrauterina se supone debido a la ausencia de la capacidad de aromatización de andrógenos (androstenediona a estrona) la placenta, impide la acumulación de androstenediona. Se han descrito 5 isoenzimas de los cuales sólo el tipo 3 es relevante, habiéndose encontrado el agregado más importante en árabes de Gaza, Brasil y Holanda.
e. Déficit de 5α-Reductasa
Este enzima convierte la testosterona a dihidrotestosterona (DHT) 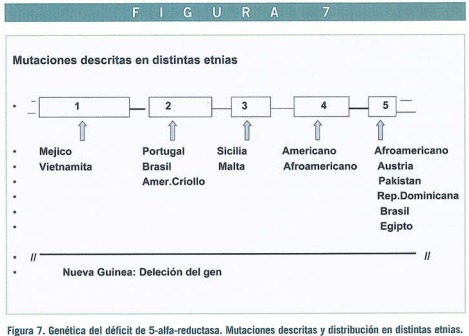 al nivel celular y su déficit intracelular conduce a un patrón de virilización de los genitales internos que responden a la testosterona durante la embriogénesis, pero el déficit de conversión a DHT, comúnmente parcial, conduce a virilización incompleta de genitales externos. Existe un amplio espectro de manifestación fenotípica aunque es característica la notable virilización de genitales externos en la pubertad. Los testículos pueden ser abdominales, inguinales o escrotales, y el diagnóstico se basa en la relación T/DHT bajo estimulación con hCG (gonadotrofina coriónica con efecto LH), puesto que los niveles basales en la infancia son excesivamente bajos para ser discriminantes. Puede confirmarse el déficit enzimático de 5α-R celular mediante cultivo de fibroblastos procedentes de células de genitales externos.
al nivel celular y su déficit intracelular conduce a un patrón de virilización de los genitales internos que responden a la testosterona durante la embriogénesis, pero el déficit de conversión a DHT, comúnmente parcial, conduce a virilización incompleta de genitales externos. Existe un amplio espectro de manifestación fenotípica aunque es característica la notable virilización de genitales externos en la pubertad. Los testículos pueden ser abdominales, inguinales o escrotales, y el diagnóstico se basa en la relación T/DHT bajo estimulación con hCG (gonadotrofina coriónica con efecto LH), puesto que los niveles basales en la infancia son excesivamente bajos para ser discriminantes. Puede confirmarse el déficit enzimático de 5α-R celular mediante cultivo de fibroblastos procedentes de células de genitales externos.
Se han tipificado 2 genes (SRD5), el tipo I localizado en el cromosoma 5 (SRD5AA) y el tipo II (SRD5A2) en el cromosoma 2 (2p23), aunque sólo el tipo II es deficiente en el seudohermafroditismo masculino, existiendo distintas mutaciones según la etnia de origen (distribuidas en los 5 exones que constituyen el gen) (Figura 7).
Insensibilidad androgénica
La forma más completa inicialmente descrita como «feminización testicular» o síndrome de Morris (Mendoca et al., 2000) se presenta en individuos 46,XY con testículo bilateral, ausencia de derivados mullerianos, genitales externos femeninos, vagina ciega y desarrollo mamario femenino. La patogenia común es el defecto en los receptores androgénicos que interfiere con la respuesta a la testosterona. La presencia de MIF (hormona testicular antimulleriana) justifica la ausencia de derivados mullerianos, pero el testículo sintetiza testosterona y estrógenos comparables al varón normal y se produce la feminización por falta del antagonismo androgénico.
Algunos individuos con insensibilidad androgénica muestran hipertrofia de clítoris y fusión labio-escrotal, considerándose como formas incompletas o parciales del síndrome, y en el extremo del espectro se encuentran formas leves con sólo ginecomastia y oligo-azoospermia. Sin embargo, el patrón de transmisión es el mismo que en las formas completas. Alrededor del 30% se consideran casos esporádicos de novo y en el resto la madre, fenotípicamente normal, o con escaso vello púbico y retraso puberal, sería heterocigótica.
Se trata de una alteración recesiva ligada al cromosoma X, y el gen que codifica el receptor androgénico (AR) está constituido por 8 exones y se localiza en el brazo largo del cromosoma X (Xq11-12). Se han descrito muchas distintas mutaciones (Gottlieb et al., 1999) recopiladas en un registro internacional de la Universidad canadiense McGill. Las mutaciones más comunes están en los exones 5-8 que es el dominio de fijación androgénica, no existiendo una correlación entre las distintas mutaciones y su expresividad más o menos completa. Las diferencias al nivel celular de las formas parciales pueden residir en una reducción en el número o en la calidad de los receptores (Pinsky et al., 1987). Las formas previamente descritas como síndromes de Reifesntein o síndrome de Lubs se consideran actualmente como formas parciales de insensibilidad androgénica. La importancia clínica en estos casos reside en que debe identificarse antes de la asignación de sexo, pudiendo descartarse mediante la valoración de la respuesta a los andrógenos.
Referencias
Achermann JC, Ito M, Hindmarsh PG, Jameson JL. A mutation in the gene encoding steroidogenic factor 1 causes XY sex reversal and adrenal failure in humans. Na. Genetics. 1999; 22:125-6
ASRM. Clasification of adnexal adhesions, distal tubal oclusions, mullerian anomalies and intrauterine adhesioons. Fertil. Steril. 1988;49:944-55
Avner P and Heard E. X-cromosome inactivation; counting, choice and initation. Nat.Rev.Genet. 2001; 2:59-67
Bione S and Toniolo D. X chromosome genes and premature ovarian failure. Sem. reprod. Med. 2000; 18:51-7.
Burgoyne PS. Genetic homology and crossing over in the X and Y chromosomes of Mammals. Human Genet. 1982; 61:85-90.
Foudila T, Sondrerstrom-Antila V, Hovatta O. Turner´s syndrome and pregnancies afer oocyte dontaion. Hum. Reprod. 1999; 14:532-5.
Gottlieb B, Pinsky L, Beitel LK. Androgen insensitivity. Am.J.Med. Gentet. 1999; 89:210.
Huang B, Wang S, Ning Y. Autosumal XX sex reversal caused by duplication of SOX9. Am. J.Med Genet. 1999; 87:349.
Kahn AK, Abdalla HI, Oskarsson T. Two successful pregnancies in a 46,XY patient. Human Reprod. 1997; 12:1434-5.
Laymann LC. Genetics of human hypogonadotrofic hypogonadism. Am.J.Hum.Genet. 1999; 89:240-8.
Lin PC, Bhatnagar KP, nettleton GS, Nakajima ST. Female genital anomalies affecting reproduction. Fertil.Steril 2002; 78:899-915.
McDonough PG and Tho PT. The spectrum of 45,X/46,XY gonadal and its implication. Pedistr.Adolesc. Gynecol. 1983; 1:-.
Meeks J, Weiss J, Jameson JL. Dax1 is required for testis determination. Nature Genetics. 2003; 34:32-33.
Mendonca BB, Inacio M, Arnohold IJ. Male seudohermafroditism due to 17b HDS deficiency. Diagnosis, psychological evaluation and Management. Medicine 2000. 79:299-309.
Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al., Congenital leptin defiency is associated with severe early onset obesity in humans. Nature. 1997; 387:903-8.
Morris JM Mahest VB. Further observations on the syndrome testicular feminization. Am.J.Obstet.Gynecol.1963; 87:731-48.
Ohno S. Sex chromosomes and sex linked genes. Springer. Berlin. 1967.
Pinsky L, Kaufma M, Levitsky LL. Partial androgen resistance due to a distinctive qualitative defect of the androgen receptor. Am.J.Med.Genet. 1987; 27:459-66.
Pru JK y Tilly JL, Programmed cell death in the ovary. Insights and future prospects using genetic technologies. Mol. Endocrinol. 2001; 15:845-53.
Rosenberg C, Frota-Pessoa O, Viana Morgante AM, Chu TH. Phenotypic spectrum of 45,X/46,XY individuals. Am.J.Med.Genet. 1987; 27: 553-9.
Rosenfeld RG, Attie KM, Frane J, Brasel JA, Burstein S, Cara JF, et al., Growth hormone therapy of Turner´s syndrome: beneficial efect on adult height. J.Pediatr. 1998; 132, 319-24.
Rosenfeld RG, Grumbach MM. Turner Syndrome. Ed. Marcel Dekker 1190. New York.
Simpson JL. Disorders of sexual diferentiation. NY Academic Press. 1976.
Simpson JL. Pregnancies in women with chromosomal anomalies. En Genetic Diseases in pregnancy. 1981. New York. Academic Press.
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, et al. The male specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003; 423:825-837.
Sultan LH and Lumbroso S. LH receptor defects. En Fertility and Reproductive Medicine. Proc. XVI World Congress on Fertility and Sterility. Elsevier. 1998. Amsterdam.
Verp MS, Harrison HH, Ober C, Oliveri D, Amarose AP, Lindgren V, et al. Chimerism as the etiology of a 46,XX/46,XY fertile true hermafrodite. Fertil. Steril. 1992; 57: 346-9.