EPIGENÉTICA EN TÉCNICAS DE REPRODUCCIÓN ASISTIDA: RAZONES Y EVIDENCIAS PARA UNA REFLEXIÓN
Ariadna Brotons1,2*, Ana A Fernández-Peinado1, Antonio Moya1, Paz Ibañez1, Francisco J Vidal-Iglesias2, José Solla-Gullón2, Jesús Iniesta2
1In Vitam Centro de Medicina Reproductiva, Avenida Universidad, 24, 03202, Elche, Alicante, España.
2Instituto Universitario de Electroquímica, Universidad de Alicante, Carretera de San Vicente del Raspeig s/n, 03080, Alicante, España.
Los procesos epigenéticos producen cambios covalentes en la cadena de ADN sin alteración en la secuencia de bases nitrogenadas. Una de las modificaciones epigenéticas más relevantes es el proceso de metilación de citosina. Este proceso es de vital importancia para mantener el silenciamiento génico en el desarrollo normal, la impronta genómica y la inactivación del cromosoma X. Así, por ejemplo, cuando se producen alteraciones en la impronta, estos pueden desencadenar enfermedades, especialmente aquellas relacionadas con defectos en el desarrollo embrionario y el proceso neoplásico. En los últimos años se ha visto que mediante el uso de técnicas de reproducción asistida (TRAs), se pueden ver aumentados los desórdenes de este tipo, motivación suficiente por la que creemos que resulta de interés un mayor conocimiento de las modificaciones epigenéticas y su relación con numerosas enfermedades y disfunciones tras el uso de TRAs, con notables consecuencias sobre el manejo, tratamiento y prevención en el futuro de enfermedades en recién nacidos. En este trabajo, se describen los mecanismos de las modificaciones epigenéticas, más concretamente la metilación de citosina, las implicaciones que tiene dicha metilación de ADN sobre disfunciones y desarrollo de enfermedades en recién nacidos, los factores implicados en TRAs causantes y/o transmisión de la metilación de ADN y, finalmente, las metodologías analíticas para la determinación del grado de metilación en ADN.
Entre un 10 y un 15% de la población de la sociedad moderna padece problemas de esterilidad/infertilidad, por lo que cada vez son más utilizadas las técnicas de reproducción asistida (TRA), y el número de niños nacidos gracias a ellas ha aumentado. Son muchos los estudios que aseguran que, tras el empleo de estas técnicas, existe un incremento del riesgo de crecimiento intrauterino restringido, parto pretérmino (Oakes CC et al., 2007), muerte fetal intrauterina (Ziller MJ et al., 2011), alteraciones de los cromosomas sexuales (Li JY et al., 2004), cardiopatías (Hata K et al., 2002), síndrome de Beckwith-Wiedemann (Davis TL et al., 2000) y síndrome de Angelman (Ludwig M et al., 2005), aunque también existen autores que aseguran que aún no son suficientes las evidencias que confirmen una estrecha relación de las TRA con estos dos síndromes (Li JY et al., 2004). Las posibles causas del incremento de estas patologías, en comparación con los recién nacidos procedentes de embarazos espontáneos, parecen estar asociadas a alteraciones ya existentes en los gametos de los padres o, por otra parte, a factores directamente derivados de las TRA. A lo largo de los años las causas cromosómicas se han estudiado ampliamente, sin embargo, las modificaciones en la expresión de genes que no se encuentran en la secuencia del ADN, conocidas como modificaciones epigenéticas (herencia epigenética) han sido menos estudiadas. En este artículo intentaremos ampliar este concepto y analizar en detalle las posibles consecuencias que estas alteraciones del ADN pueden producir en los futuros niños nacidos gracias a las TRA.
Epigenética
Los cambios epigenéticos son modificaciones covalentes de la cadena de ADN que no originan cambios en la secuencia de bases nitrogenadas (Wolffe AP y Matzke MA, 1999). La epigenética, por ejemplo, nos permite explicar la diferencia existente entre el ser humano y el chimpancé a pesar de compartir el 99% de los genes. Los tres principales tipos de información epigenética son: modificación de histonas, metilación de la citosina e impronta genética.
Modificación de histonas:
Las histonas pueden ser modificadas tras la traducción, lo que cambia sus propiedades de unión al ADN y a proteínas nucleares. Estas modificaciones, que pueden ser heredadas, influyen en la expresión génica y cambian la arquitectura local de la cromatina. Entre las modificaciones covalentes más importantes que pueden sufrir las histonas encontramos la acetilación, la metilación y la fosforilación. Hoy en día parece claro que determinados patrones de modificación de histonas conducen a determinadas patologías entre las que podrían encontrarse, además de patologías tumorales, patologías inflamatorias de localización broncopulmonar como el asma.
Metilación de ADN
La metilación, descrita por primera vez en los años 70 (Holliday R y Pugh JE, 1975), es una modificación del ADN, en la que un grupo metilo es transferido desde la S-adenosilmetionina a una posición C-5 de citosina por una ADN-5 metiltrasferasa (DNMT) (Laird PW, 2003) dando lugar a la formación de la metilcitosina (mC), como se muestra en la Figura 1.

En las células somáticas humanas, las mC constituyen el 1% del total de las bases del ADN y en la mayoría de los casos se presentan en forma de dinucleótidos -CpG-. Estos dinucleótidos no están distribuidos de forma uniforme, sino que en el 98% del genoma los CpG están presentes en un promedio de uno por cada 80 dinucleótidos, existiendo regiones de 200 pb a varias kilobases que tienen una frecuencia cinco veces mayor de dinucleótidos CpG, llamadas “islas CpG”. En general, estas islas se localizan en la región central del promotor y el sitio de inicio de la transcripción, observándose represión en la expresión del gen cuando se encuentran hipermetiladas (Rodríguez-Dorantes M et al., 2004). En los últimos años también se ha observado metilación de las citosinas en células embrionarias en dinucleótidos CpA y CpT (Lister R y Ecker JR, 2009, Ziller MJ et al., 2011).
Existen dos tipos de metilación. La primera corresponde a la metilación de mantenimiento, que añade grupos metilo a cadenas de ADN en lugares opuestos a los metilados en la cadena madre, provocando que las moléculas hijas de ADN mantengan un patrón de metilación después de la división celular. La segunda corresponde a la metilación de novo, que añade grupos metilo en posiciones totalmente nuevas, pudiendo cambiar el patrón de metilación en una región localizada del genoma.
Inicialmente se pensó que las células normales estaban sin metilar, a excepción de los genes asociados a genes improntados y genes en el cromosoma X inactivados. Actualmente, se sabe que algunos de estos islotes están metilados en células normales y que este mecanismo tiene un importante papel en la regulación de la expresión del gen. En general, un gen metilado es inactivo y los genes que se transcriben activamente poseen islotes CpG sin metilar (o hipometilados), por lo que la metilación de la citosina puede desempeñar un papel en la regulación de la expresión génica.
En genes cuyos islotes CpG no están metilados, la transcripción se inicia libremente en presencia de los factores adecuados (Figura 2A), pero la metilación puede inactivar la transcripción por varios mecanismos: impidiendo la unión de factores de transcripción (Figura 2B), o siendo reconocidos por proteínas (MeCp1, MeCp2) que se unen bloqueando la unión de los factores de transcripción (Figura 2C) o la acción de la polimerasa (Figura 2D).
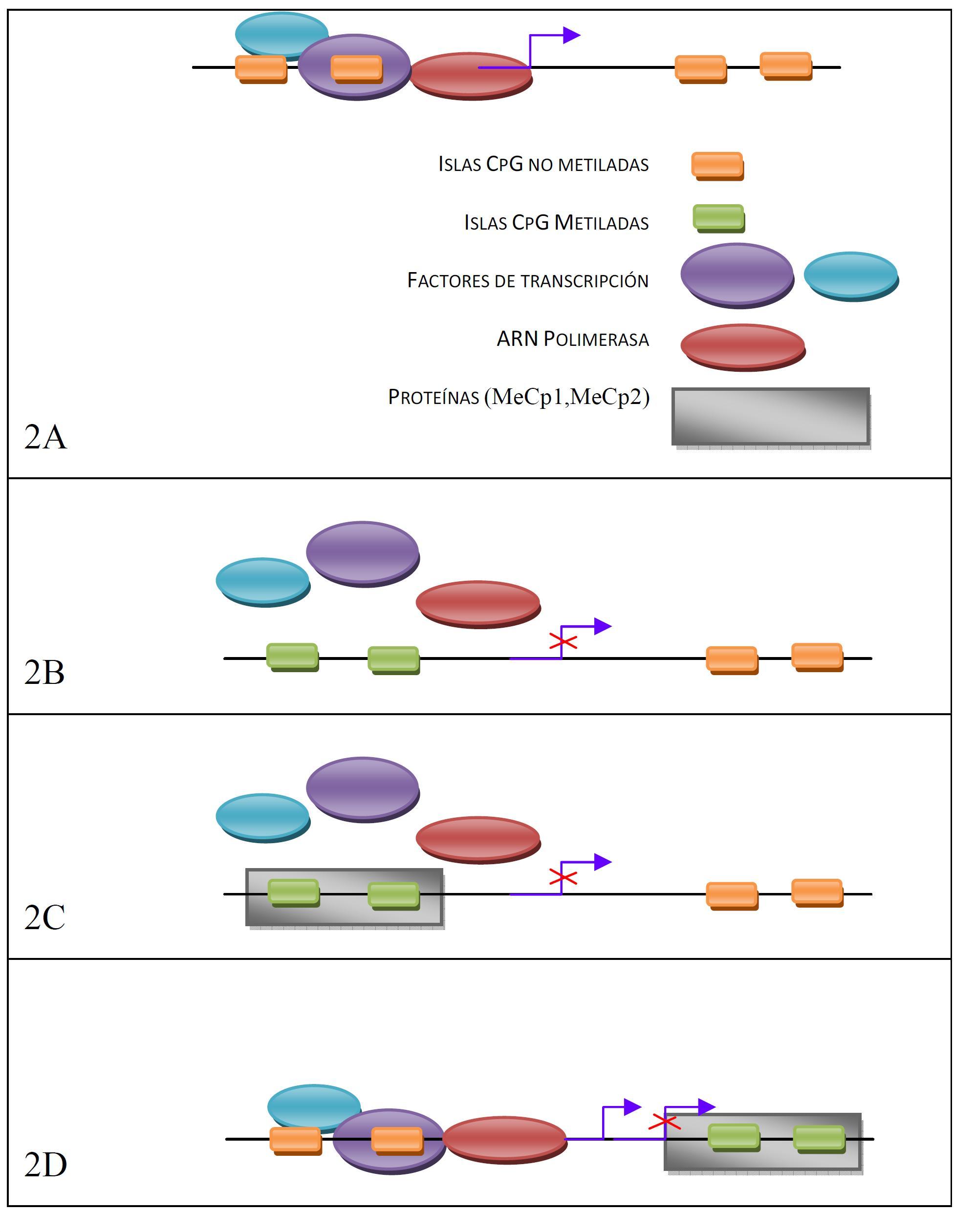
La metilación en algunas de las islas CpG en tejidos no malignos aumenta con la edad (Ahuja N et al., 1998), pero el grado de metilcitosinas en contenido total del genoma disminuye (Hoal-van Helden EG y van Helden PD, 1989). Cuando la metilación de novo se da en células somáticas adultas (debido al envejecimiento o a procesos neoplásicos) ocurre de forma muy lenta. No obstante, se ha observado que hay patrones de metilación anormales en muchos tipos de cáncer, los cuales producen principalmente inactivación de genes supresores de tumores e inestabilidad del genoma (Bird AP y Wolffe AP, 1999). Aunque no tenemos que olvidar que los patrones de metilación son vitales para el desarrollo normal de los vertebrados (Li E et al., 1992, Okano M et al., 1999), tanto el aumento como el defecto de metilación pueden ocasionar problemas graves.
También llamada impronta genómica o gamética, se manifiesta en la minoría de genes en los mamíferos (0.1-1%). Los organismos diploides poseen dos copias de cada gen autosómico, cada uno de ellos procedente de cada parental. La mayoría de los genes autosómicos expresan de forma simultánea ambos alelos, pero en un grupo pequeño de genes, uno de los alelos es silenciado por un proceso de imprinting (Manipalviratn S et al., 2009), es decir que los genomas maternos y paternos no son funcionalmente equivalentes en estos loci (Pérez Jurado LA, 2004). Parece ser que existe un mecanismo celular que de algún modo «marca» o deja una impronta sobre todos los genes «improntables» de acuerdo al sexo del individuo. De momento se conocen unos 80 genes improntados en los mamíferos y se han confirmado unos 40 en los humanos (Morison IM et al., 2005). Estos genes tienen funciones en el control del desarrollo embrionario placentario y en la supresión de tumores (Moll AC et al., 2003) y ya se han detectado 10 síndromes causados por errores en la impronta. (Manipalviratn S et al., 2009).
En el caso de la impronta, la metilación se establece en la línea germinal durante la gametogénesis y se mantiene sin cambios a lo largo del desarrollo embrionario y postnatal. El establecimiento de estos patrones de metilación contrastantes provoca que, tras la fecundación, ciertos genes sean activos en uno de los cromosomas parentales, pero no en el otro. La consecuencia a nivel genético es la presencia de un único alelo para el gen improntado, lo cual tiene implicaciones clínicas cuando hay fallos en la impronta. De hecho, esta diferencia en la metilación de los pronúcleos femenino o masculino hace que sea imposible obtener zigotos a partir de los pronúcleos, ya que esto daría lugar a la ausencia de alelos activos para determinados genes (Barton SC et al., 1984)
Aunque no se conoce a fondo, se sabe que el proceso mediante el cual estos genes adquieren impronta genómica es muy parecido al proceso de borrado y regrabación de cualquier soporte de memoria. Este proceso de regrabado es extremadamente importante, ya que las improntas han de transmitirse a la siguiente generación. Todos contamos con un juego de cromosomas con improntas procedentes del padre y otro juego de cromosomas procedentes de la madre. Pasar estos cromosomas a la siguiente generación implica reprogramar totalmente las improntas en la línea germinal para formar gametos, ya que los gametos masculinos deberán aportar únicamente improntas masculinas y para los femeninos deberá ocurrir lo mismo. Por lo tanto, durante la formación de los gametos, habrá que hacer una limpieza de improntas del sexo contrario.
La reprogramación transcurre de la siguiente manera, de acuerdo con la Figura 3. En primer lugar, en la línea germinal se eliminan las metilaciones parentales, que se completa durante los días 12-13 en células de ratón, tanto de hembras como de machos (Reik W y Dean W, 2001). Tras el borrado de las marcas de imprinting, se reestablecen las marcas de novo de acuerdo con el sexo. El proceso de restablecimiento comienza en las líneas germinales de ambos sexos en los estadios fetales tardíos y continúa hasta el nacimiento (Thonneau P et al., 1991). En las células germinales femeninas (ovocitos), el restablecimiento del imprinting se produce durante el crecimiento y no se termina hasta la ovulación de cada ciclo menstrual, repitiéndose durante toda la vida reproductiva de la mujer (Boissonnas CC et al., 2013). En el caso de las células germinales masculinas se termina el proceso de imprinting durante la gametogénesis y durante el estadio de espermátide (De Kretser DM, 1997). En el modelo de ratón, se produce una desmetilación entre los días 8 y 13.5 post coito, provocando el borrado de todas las marcas de metilación (Hajkova P et al., 2008). La remetilación comienza antes del nacimiento (15.5-18.5 días postcoito) tanto en las hembras como en los machos, pero no se completa hasta el nacimiento (Li JY et al., 2004) Por el contrario, la metilación de novo se completa durante la fase final de maduración, justo antes o durante la ovulación de los ovocitos (Dohle GR et al., 2002, Simoni M et al., 1997).
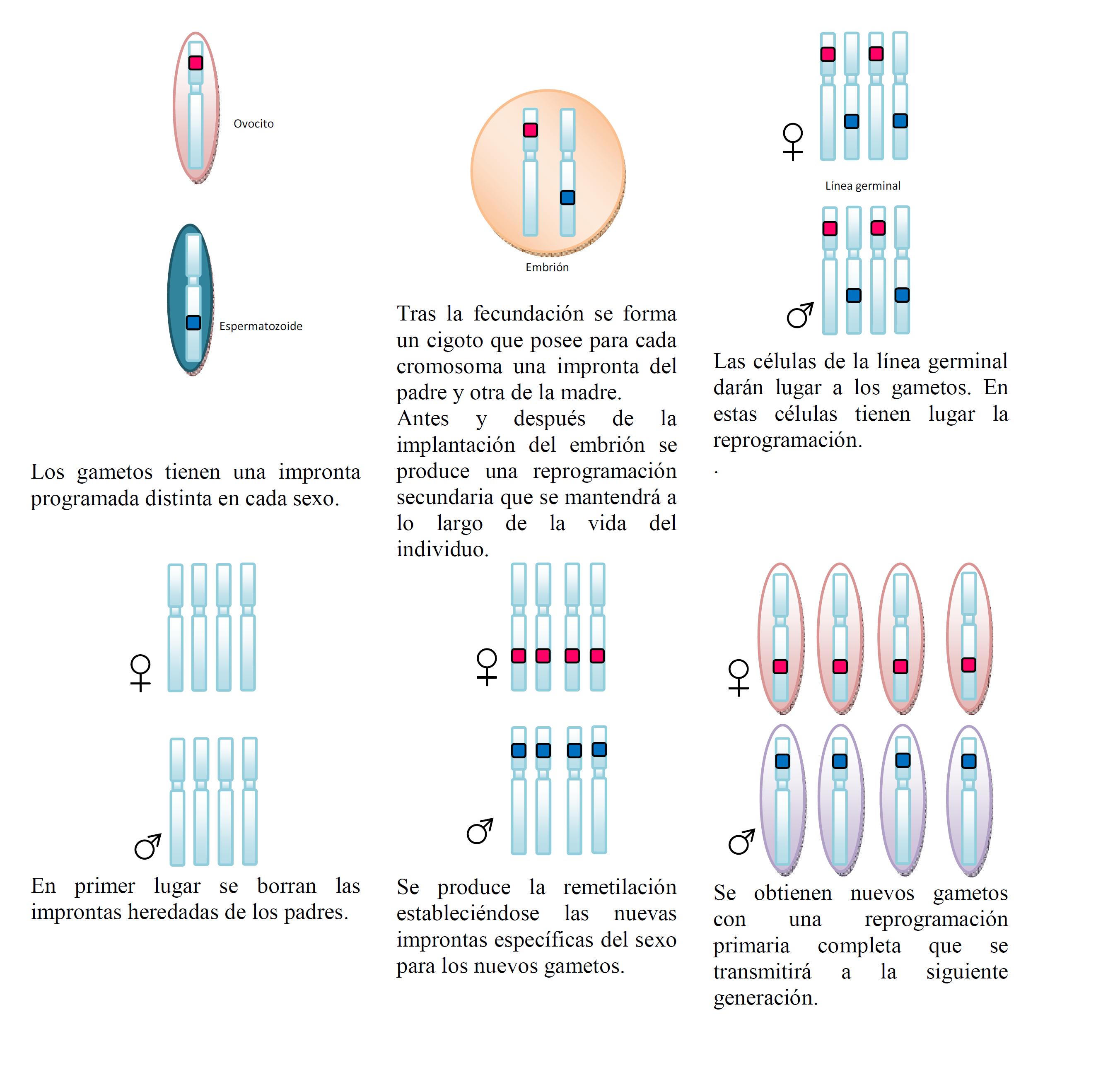
Casi todo el conocimiento que tenemos sobre el imprinting de células germinales es en el modelo de ratón, siendo escasos los estudios en células germinales humanas. Existen dos estudios que confirman la metilación del H19 de forma diferencial entre las espermatogonias y los espermatocitos tipo I (Joana Marques C et al., 2011). Hasta ahora se ha considerado que en el modelo humano pasa algo parecido al de ratón, de modo que las marcas de metilación se deben adquirir antes de entrar en meiosis, pero no se sabe si esto ocurre durante el desarrollo fetal, en el periodo perinatal y/o durante la pubertad.
Relación entre las alteraciones epigenéticas y las tras.
Aunque el papel de la metilación del ADN no está totalmente entendido, sabemos que una de sus funciones principales es el control de la expresión genética y la protección del organismo frente a la expresión de secuencias indeseadas (Havlis J y Trbusek M, 2002). Sin embargo, la hipermetilación de las islas CpG está asociada a algunas enfermedades. Por ejemplo, está asociada con la inactivación de la transcripción del gen supresor de tumores, lo cual observamos en carcinomas (Parry L y Clarke AR, 2011), leucemia (Chen J et al., 2010), cáncer de pulmón (Belinsky SA et al., 1998), tiroides (Russo D et al., 2011) páncreas (Delpu Y et al., 2011) y próstata (Donkena KV et al., 2010), entre otros muchos. También encontramos fallos de metilación en casos de esterilidad masculina (Navarro-Costa P et al., 2010, Wu W et al., 2010) y en algunos desórdenes como los síndromes Beckwith-Wiedemann y Angelman en recién nacidos concebidos mediante TRA (Maher ER et al., 2003, Owen CM y Segars JH, Jr., 2009). Esto puede ser debido a la manipulación existente en muchas de las etapas de la fecundación, incluyendo: utilización de hormonas para la estimulación ovárica, maduración in vitro de ovocitos, uso de espermatozoides inmaduros, microinyección de espermatozoides, cultivo in vitro de embriones, crioconservación de gametos y crioconservación de embriones. Cualquiera de estos procesos puede alterar el imprinting, pero debido a los problemas éticos que supone trabajar con embriones humanos, la mayor parte de los datos que se poseen provienen de modelos animales. Algunas de las evidencias existentes entre la relación de los desórdenes de la impronta y las TRA las enumeramos a continuación:
Esterilidad masculina: En el caso de las causas de esterilidad masculina, durante años se han estudiado diferentes aspectos como las anormalidades en el cariotipo (Dohle GR et al., 2002) y microdeleciones del cromosoma Y (Simoni M et al., 1997), pero las causas epigenéticas se han mantenido al margen en los estudios, y se desconocen aún sus consecuencias, aunque ya existen evidencias en la bibliografía que relacionan los marcadores epigenéticos y la fertilidad masculina. En estudios realizados con ratones a los cuales se les administró un agente desmetilante (5-aza-2’-deoxycytidine), se observó una disminución en la producción de esperma, tamaño del teste, epidídimo y aumento de mortalidad neonatal (Doerksen T y Trasler JM, 1996, Kelly TLJ et al., 2003). También se han relacionado niveles altos de metilación en el ADN espermático con tasas altas de embarazo y los defectos de metilación con infertilidad (Benchaib M et al., 2005). Otros trabajos ven en las muestras de baja calidad un elevado grado de metilación en genes imprintados y no imprintados (Houshdaran S et al., 2007). Por tanto, se ha propuesto que el elevado grado de metilación se debe al borrado incompleto de las marcas de metilación en casos de oligoastenoteratozoospermia, más que debido a errores en la metilación de novo. Además, se han asociado defectos de metilación espermática con la fertilidad (Aston KI et al., 2012). Otros autores han comparado los genes H19DMR metilado y MESTDMR no metilado en espermatozoides de varones fértiles e infértiles y relacionaron una pérdida de metilación del H19 DMR con oligoastenoteratozoospermia (OTA) (Marques CJ et al., 2004). También se ha asociado la pérdida de metilación en H19, GTL2 y cambios en el patrón de metilación en PEG1, LIT1, ZAC, PEG3 y SNRPN con oligozoospermia severa (Kobayashi H et al., 2007).
Embriogénesis: La embriogénesis no se puede dar de forma adecuada sin el mecanismo correcto de regulación epigenética. Se han realizado experimentos con ratón y se ha observado que se producen abortos cuando se bloquea la expresión de la metiltransferasa o se modifican las histonas (Benchaib M et al., 2005, Bourc’His D y Bestor TH, 2004, Hata K et al., 2002, Kerjean A et al., 2000, Li E et al., 1992, Okano M et al., 1999). También se ha visto que utilizando el modelo de ratón es necesaria tanto la contribución cromosómica materna como paterna para el desarrollo normal (McGrath J y Solter D, 1984, Surani MAH et al., 1984).
Cultivo embrionario: Un estudio reciente ha confirmado que los embriones de ratón en dos células tienen un grado de metilación superior a los desarrollados in vivo. En el mismo estudio se apreció un mayor grado de desmetilación en los pronúcleos masculinos de los embriones cultivados in vitro comparado con aquéllos in vivo, mientras que en los pronúcleos femeninos no se encontraron diferencias (Zaitseva I et al., 2007). Este hallazgo soporta la hipótesis de que el cultivo embrionario puede ser el responsable de la hipermetilación hallada en los embriones (Niemann H y Wrenzycki C, 2000). Otro estudio en ratones ha demostrado una expresión diferencial y un imprinting distinto en el gen H19 usando dos medios de cultivo diferentes, a pesar de que la ADN metiltransferasa y la distribución de la proteína DNMT1 era similar en ambos grupos (Doherty AS et al., 2000). Por el contrario, en otro estudio también en ratón, se muestra un grado distinto de expresión en distintos genes improntados, incluyendo H19, IGF2, GRB10 y GRB7 en embriones cultivados in vivo, ex vivo en medio M16 y ex vivo en medio M6 mas suero bovino fetal (Khosla S et al., 2001). También se ha observado que los embriones en 2 células poseen menos alteraciones de la metilación que los blastocistos, aumentándose las aberraciones en un mayor número de genes y observándolas tanto en tejido embrionario como extraembrionario (Rivera RM et al., 2008). Todos estos estudios parecen reforzar la hipótesis de que el medio de cultivo afecta a la impronta. Algunos mecanismos por los cuales los medios de cultivo pueden afectar en la impronta son: (1) fallo del movimiento de la DNMT1 hacia el interior del núcleo en el estadio correcto del desarrollo embrionario (Thompson JR y Williams CJ, 2005), (2) alteración de la expresión de la DNMT y otras proteínas que intervienen en el imprinting (Thompson JR y Williams CJ, 2005), (3) alteración del ciclo celular en el momento de la reprogramación epigenética (De Rycke M et al., 2002, Thompson JR y Williams CJ, 2005) e (4) interacción del grupo metilo con el medio de cultivo (De Rycke M et al., 2002).
Estimulación ovárica: Siguiendo con el modelo en ratón, se ha observado que los embriones obtenidos de ovocitos tras estimulación hormonal presentan más alteraciones de la metilación comparados con los obtenidos sin estimulación (Shi W y Haaf T, 2002). Para evitar esto, algunos autores han propuesto el uso de la maduración in vitro de ovocitos, pero en estudios con ovocitos bovinos se han encontrado también alteraciones de la metilación tras maduración in vitro (Hiendleder S et al., 2006). El número de ovocitos estudiados es muy pequeño, pero parece que los defectos de metilación son menores cuando se utiliza esta técnica (de un estudio de 20 ovocitos, se encontraron defectos en sólo 5) (Borghol N et al., 2006).
Abortos: Existen trabajos que relacionan abortos espontáneos con las mismas alteraciones de metilación que se encuentran en los espermatozoides tras la utilización de TRA (Kobayashi H et al., 2009, Kobayashi H et al., 2007).
Consecuencias de las alteraciones epigenéticas tras TRAs
A continuación citamos algunos síndromes que están asociados con el mayor o menor grado de metilación en genes humanos y animales y de los que se ha observado un aumento en niños nacidos tras TRA.
Síndrome de la cría grande (Large offspring syndrome)
Este síndrome se caracteriza por sobrepeso en el nacimiento, distrés respiratorio, hiperplasia de los órganos, anomalías esqueléticas, y un aumento de muerte súbita (Young LE et al., 1998). Se relaciona con una disminución de la metilación y de la expresión del gen IGF2R, lo cual ocurre en ovejas tras TRA con cocultivo de células de la granulosa y/o suero (Young LE et al., 2001).
Síndrome de Beckwith-Wiedemann
El síndrome de Beckwith-Wiedemann es un desorden congénito que implica crecimiento exagerado hiperplasia-hipertrófica. Los primeros indicios de la enfermedad son macrosomía (elevado peso al nacer), macroglosia (lengua grande), defectos de la pared media abdominal como onfalocele (los pacientes presentan de forma típica vísceras fuera de la zona abdominal) y predisposición a cáncer embrionario (DeBaun MR et al., 2003). Este síndrome está causado por mutaciones en los genes reguladores del crecimiento situados en el cromosoma 11 (en la región 11p15.5) o por errores en impronta genómica. La zona imprintada del cromosoma 11 incluye dos subdominios: uno más centromérico que incluye en p57KIP2, LIT, TSSC3 y TSSC5 y otro más telomérico que incluye IGF2 y H19 (Lee J et al., 2002). Existen diversas publicaciones donde se confirma el aumento de casos de niños nacidos con este síndrome cuando se utilizan TRA (Gicquel C et al., 2003, Halliday J et al., 2004, Maher ER et al., 2003) (de un 1.2% de la población general a un 4% en los nacidos tras TRA (Maher ER et al., 2003)).
Síndrome de Angelman
El síndrome de Angelman se caracteriza por un retraso en el desarrollo cognitivo, una capacidad lingüística reducida o nula, escasa receptividad comunicativa, escasa coordinación motriz, con problemas de equilibrio y movimiento, ataxia, estado aparente permanentemente de alegría, con risas y sonrisas en todo momento, siendo fácilmente excitables, hipermotricidad y déficit de atención. Es un ejemplo clásico de enfermedad con herencia epigenética, dado que las mutaciones y defectos que lo causan podrían estar implicadas en el desarrollo de la enfermedad en función de si la copia del gen alterado proviene del padre o de la madre. La zona donde se encuentran estas mutaciones está en el cromosoma 15, en el loci 15q11-q13 (Cox GF et al., 2002). Desafortunadamente, no hay datos claros sobre cuál es el aumento de este síndrome en niños nacidos por TRA, pero sí que se ha podido observar que el 20% de niños con este síndrome proceden de parejas subfértiles (Ludwig M et al., 2005).
Retinoblastoma
El retinoblastoma es un cáncer de la retina causado por una mutación en la proteína Rb, codificada por un gen supresor tumoral denominado RB1, en el cromosoma 13q14. En algunos casos este gen se silencia epigenéticamente mediante la hipermetilación del promotor situado en el exón 1 (Greger V et al., 1989). Este tumor se produce fundamentalmente en niños pequeños y representa el 3% de los cánceres en menores de quince años. Constituye la primera causa de malignidad intraocular primaria en los niños, por lo que la incidencia anual estimada es del 1-1.5% de la población; sin embargo, entre los nacidos mediante TRA esta cifra asciende y ronda el 4.9 y 7.2 %.
Determinación de la metilación del ADN.
La importancia en la determinación del grado de metilación del ADN ha hecho que se pongan a punto diversas técnicas con dicho fin como el Bisulfito (Frommer M et al., 1992), PCR (Kane MF et al., 1997), MSP (Methylation-specific PCR) (Herman JG et al., 1996), MS-SNuPE (methilation-sensitive single nucleotide primer extension) (Kuppuswamy MN et al., 1991), y microarrays (Adorján P et al., 2002), entre otras. Las metodologías citadas anteriormente son potentes para el estudio de la metilación del ADN pero técnicamente laboriosas, largas, poco sensibles, caras y no lo suficientemente reproducibles. En los últimos años se están desarrollando diferentes biosensores electroquímicos (Brett AMO et al., 2000, Brotons A et al., 2013, Kato D et al., 2011) que permiten realizar diferentes ensayos más sencillos, baratos y rápidos, con el fin de implantarlos en los laboratorios de FIV con aplicaciones diagnósticas. La bibliografía en este campo es numerosa, por lo que no es el objeto de este artículo una revisión exhaustiva de las diferentes líneas de investigación sobre el uso de biosensores electroquímicos para la determinación del grado de metilación en ADN.
conclusiones
El conocimiento de las modificaciones epigenéticas que ocurren en las enfermedades humanas es de vital importancia para abordar el tratamiento de las mismas en el futuro. Los cambios en los patrones de metilación podrán ser empleados como biomarcadores en cáncer y en reproducción asistida, entre otras enfermedades o disfunciones. El estado potencialmente reversible de este proceso constituye un blanco ideal para crear estrategias terapéuticas que impliquen la reactivación o el re-silenciamiento de genes específicos, por lo que debemos seguir investigando en este campo y desarrollando técnicas que faciliten la detección y análisis del grado de avance de dichas alteraciones.
Referencias
Adorján P, Distler J, Lipscher E, Model F, Müller J, Pelet C, et al. Tumour class prediction and discovery by microarray-based DNA methylation analysis. Nucleic Acids Res 2002; 30:e21
Ahuja N, Li Q, Mohan AL, Baylin SB y Issa JPJ. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998; 58:5489-5494
Aston KI, Punj V, Liu L y Carrell DT. Genome-wide sperm deoxyribonucleic acid methylation is altered in some men with abnormal chromatin packaging or poor in vitro fertilization embryogenesis. Fertil Steril 2012; 97:285-292.e284
Barton SC, Surani MAH y Norris ML. Role of paternal and maternal genomes in mouse development. Nature 1984; 311:374-376
Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, et al. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A. 1998; 95:11891-11896
Benchaib M, Braun V, Ressnikof D, Lornage J, Durand P, Niveleau A, et al. Influence of global sperm DNA methylation on IVF results. Hum Reprod 2005; 20:768-773
Bird AP y Wolffe AP. Methylation-induced repression-belts, braces, and chromatin. Cell 1999; 99:451-454
Boissonnas CC, Jouannet P y Jammes H. Epigenetic disorders and male subfertility. Fertil Steril 2013; 99:624-631
Borghol N, Lornage J, Blachère T, Sophie Garret A y Lefèvre A. Epigenetic status of the H19 locus in human oocytes following in vitro maturation. Genomics 2006; 87:417-426
Bourc'His D y Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 2004; 430:96-99
Brett AMO, Piedade JAP y Serrano SHP. Electrochemical oxidation of 8-oxoguanine. Electroanalysis 2000; 12:969-973
Brotons A, Mas LA, Metters JP, Banks CE y Iniesta J. Voltammetric behaviour of free DNA bases, methylcytosine and oligonucleotides at disposable screen printed graphite electrode platforms. Analyst 2013; 138:5239-5249
Cox GF, Bürger J, Lip V, Mau UA, Sperling K, Wu BL, et al. Intracytoplasmic sperm injection may increase, the risk of imprinting defects. Am J Hum Genet 2002; 71:162-164
Chen J, Odenike O y Rowley JD. Leukaemogenesis: more than mutant genes Nat Rev Cancer 2010; 10:23
Davis TL, Yang GJ, McCarrey JR y Bartolomei MS. The H19 methylation imprint is erased and re-established differentially on the parental alleles during male germ cell development. Hum Mol Genet 2000; 9:2885-2894
De Kretser DM. Male infertility. Lancet 1997; 349:787-790
De Rycke M, Liebaers I y Van Steirteghem A. Epigenetic risks related to assisted reproductive technologies: Risk analysis and epigenetic inheritance. Hum Reprod 2002; 17:2487-2494
DeBaun MR, Niemitz EL y Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet 2003; 72:156-160
Delpu Y, Hanoun N, Lulka H, Sicard F, Selves J, Buscail L, et al. Genetic and Epigenetic Alterations in Pancreatic Carcinogenesis. Curr Genet 2011; 12:15-24
Doerksen T y Trasler JM. Developmental exposure of male germ cells to 5-azacytidine results in abnormal preimplantation development in rats. Biol Reprod 1996; 55:1155-1162
Doherty AS, Mann MRW, Tremblay KD, Bartolomei MS y Schultz RM. Differential effects of culture on imprinted H19 expression in the preimplantation mouse embryo. Biol Reprod 2000; 62:1526-1535
Dohle GR, Halley DJJ, Van Hemel JO, Van Den Ouwel AMW, Pieters MHEC, Weber RFA, et al. Genetic risk factors in infertile men with severe oligozoospermia and azoospermia. Hum Reprod 2002; 17:13-16
Donkena KV, Young CYF y Tindall DJ. Oxidative stress and DNA methylation in prostate cancer. Obstet Gynecol 2010; 2010:302051-302051
Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. A genomic sequencing protocol that yields a positive display of 5- methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992; 89:1827-1831
Gicquel C, Gaston V, Mandelbaum J, Siffroi JP, Flahault A y Le Bouc Y. In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCNQ1OT gene [1]. Am J Hum Genet 2003; 72:1338-1341
Greger V, Passarge E, Hopping W, Messmer E y Horsthemke B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet 1989; 83:155-158
Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 2008; 452:877-881
Halliday J, Oke K, Breheny S, Algar E y Amor DJ. Beckwith-Wiedemann syndrome and IVF: A case-control study [4]. Am J Hum Genet 2004; 75:526-528
Hata K, Okano M, Lei H y Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 2002; 129:1983-1993
Havlis J y Trbusek M. 5-Methylcytosine as a marker for the monitoring of DNA methylation. J Chromatogr B Biomed Sci Appl 2002; 781:373-392
Herman JG, Graff JR, Myöhänen S, Nelkin BD y Baylin SB. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996; 93:9821-9826
Hiendleder S, Wirtz M, Mund C, Klempt M, Reichenbach HD, Stojkovic M, et al. Tissue-specific effects of in vitro fertilization procedures on genomic cytosine methylation levels in overgrown and normal sized bovine fetuses. Biol Reprod 2006; 75:17-23
Hoal-van Helden EG y van Helden PD. Age-related methylation changes in DNA may reflect the proliferative potential of organs. Mutat Res 1989; 219:263-266
Holliday R y Pugh JE. DNA modification mechanisms and gene activity during development. Developmental clocks may depend on the enzymic modification of specific bases in repeated DNA sequences. Science 1975; 187:226-232
Houshdaran S, Cortessis VK, Siegmund K, Yang A, Laird PW y Sokol RZ. Widespread epigenetic abnormalities suggest a broad DNA methylation erasure defect in abnormal human sperm. Plos One 2007; 2:e1289
Joana Marques C, Pinho MJ, Carvalho F, Bièche I, Barros A y Sousa M. DNA methylation imprinting marks and DNA methyltransferase expression in human spermatogenic cell stages. Epigenetics 2011; 6:1354-1361
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997; 57:808-811
Kato D, Goto K, Fujii S-i, Takatsu A, Hirono S y Niwa O. Electrochemical DNA Methylation Detection for Enzymatically Digested CpG Oligonucleotides. Anal Chem 2011; 83:7595-7599
Kelly TLJ, Li E y Trasler JM. 5-Aza-2′-Deoxycytidine Induces Alterations in Murine Spermatogenesis and Pregnancy Outcome. J Androl 2003; 24:822-830
Kerjean A, Dupont JM, Vasseur C, Le Tessier D, Cuisset L, Pàldi A, et al. Establishment of the paternal methylation imprint of the human H19 and MEST/PEG1 genes during spermatogenesis. Hum Mol Genet 2000; 9:2183-2187
Khosla S, Dean W, Brown D, Reik W y Feil R. Culture of preimplantation mouse embryos affects fetal development and the expression of imprinted genes. Biol Reprod 2001; 64:918-926
Kobayashi H, Hiura H, John RM, Sato A, Otsu E, Kobayashi N, et al. DNA methylation errors at imprinted loci after assisted conception originate in the parental sperm. Eur J Hum Genet 2009; 17:1582-1591
Kobayashi H, Sato A, Otsu E, Hiura H, Tomatsu C, Utsunomiya T, et al. Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum Mol Genet 2007; 16:2542-2551
Kuppuswamy MN, Hoffmann JW, Kasper CK, Spitzer SG, Groce SL y Bajaj SP. Single nucleotide primer extension to detect genetic diseases: Experimental application to hemophilia B (factor IX) and cystic fibrosis genes. Proc Natl Acad Sci U S A. 1991; 88:1143-1147
Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer 2003; 3:253-266
Lee J, Inoue K, Ono R, Ogonuki N, Kohda T, Kaneko-Ishino T, et al. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development 2002; 129:1807-1817
Li E, Bestor TH y Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69:915-926
Li JY, Lees-Murdock DJ, Xu GL y Walsh CP. Timing of establishment of paternal methylation imprints in the mouse. Genomics 2004; 84:952-960
Lister R y Ecker JR. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res 2009; 19:959-966
Ludwig M, Katalinic A, Groß S, Sutcliffe A, Varon R y Horsthemke B. Increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples. J Med Genet 2005; 42:289-291
Maher ER, Afnan M y Barratt CL. Epigenetic risks related to assited reproductive technologies: Epigenetics, imprinting, ART and icebergs? Hum Reprod 2003; 18:2508-2511
Maher ER, Brueton LA, Bowdin SC, Luharia A, Cooper W, Cole TR, et al. Beckwith-Wiedemann syndrome and assisted reproduction technology (ART) [5]. J Med Genet 2003; 40:62-64
Manipalviratn S, DeCherney A y Segars J. Imprinting disorders and assisted reproductive technology. Fertil Steril 2009; 91:305-315
Marques CJ, Carvalho F, Sousa M y Barros A. Genomic imprinting in disruptive spermatogenesis. Lancet 2004; 363:1700-1702
McGrath J y Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984; 37:179-183
Moll AC, Imhof SM, Cruysberg JRM, Schouten-van Meeteren AYN, Boers M y Van Leeuwen FE. Incidence of retinoblastoma in children born after in-vitro fertilisation. Lancet 2003; 361:309-310
Morison IM, Ramsay JP y Spencer HG. A census of mammalian imprinting. Trends Genet 2005; 21:457-465
Navarro-Costa P, Nogueira P, Carvalho M, Leal F, Cordeiro I, Calhaz-Jorge C, et al. Incorrect DNA methylation of the DAZL promoter CpG island associates with defective human sperm(dagger). Hum Reprod 2010; 25:2647-2654
Niemann H y Wrenzycki C. Alterations of expression of developmentally important genes in preimplantation bovine embryos by in vitro culture conditions: Implications for subsequent development. Theriogenology 2000; 53:21-34
Oakes CC, La Salle S, Smiraglia DJ, Robaire B y Trasler JM. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev Biol 2007; 307:368-379
Okano M, Bell DW, Haber DA y Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99:247-257
Owen CM y Segars JH, Jr. Imprinting Disorders and Assisted Reproductive Technology. Semin Reprod Med 2009; 27:417-428
Parry L y Clarke AR. The Roles of the Methyl-CpG Binding Proteins in Cancer. Genes cancer 2011; 2:618-630
Pérez Jurado LA. Genomic imprinting and endocrinology. Impronta genómica y endocrinología 2004; 60:49-54
Reik W y Dean W. DNA methylation and mammalian epigenetics. Electrophoresis 2001; 22:2838-2843
Rivera RM, Stein P, Weaver JR, Mager J, Schultz RM y Bartolomei MS. Manipulations of mouse embryos prior to implantation result in aberrant expression of imprinted genes on day 9.5 of development. Hum Mol Genet 2008; 17:1-14
Rodríguez-Dorantes M, Téllez-Ascencio N, Cerbón MA, Lez M y Cervantes A. DNA methylation: An epigenetic process of medical importance. Rev Invest Clin. 2004; 56:56-71
Russo D, Damante G, Puxeddu E, Durante C y Filetti S. Epigenetics of thyroid cancer and novel therapeutic targets. J Mol Endocrinol 2011; 46:R73-R81
Shi W y Haaf T. Aberrant methylation patterns at the two-cell stage as an indicator of early developmental failure. Mol Reprod Dev 2002; 63:329-334
Simoni M, Gromoll J, Dworniczak B, Rolf C, Abshagen K, Kamischke A, et al. Screening for deletions of the Y chromosome involving the DAZ (Deleted in AZoospermia) gene in azoospermia and severe oligozoospermia. Fertil Steril 1997; 67:542-547
Surani MAH, Barton SC y Norris ML. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984; 308:548-550
Thompson JR y Williams CJ. Genomic imprinting and assisted reproductive technology: Connections and potential risks. Semin Reprod Med 2005; 23:285-295
Thonneau P, Marchand S, Tallec A, Ferial ML, Ducot B, Lansac J, et al. Incidence and main causes of infertility in a resident population (1 850 000) of three French regions (1988-1989). Hum Reprod 1991; 6:811-816
Wolffe AP y Matzke MA. Epigenetics: Regulation through repression. Science 1999; 286:481-486
Wu W, Shen O, Qin Y, Niu X, Lu C, Xia Y, et al. Idiopathic Male Infertility Is Strongly Associated with Aberrant Promoter Methylation of Methylenetetrahydrofolate Reductase (MTHFR). Plos One 2010; 5:e13884
Young LE, Fernandes K, McEvoy TG, Butterwith SC, Gutierrez CG, Carolan C, et al. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat Genet 2001; 27:153-154
Young LE, Sinclair KD y Wilmut I. Large offspring syndrome in cattle and sheep. Rev Reprod 1998; 3:155-163
Zaitseva I, Zaitsev S, Alenina N, Bader M y Krivokharchenko A. Dynamics of DNA-demethylation in early mouse and rat embryos developed in vivo and in vitro. Mol Reprod Dev 2007; 74:1255-1261
Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, Bock C, et al. Genomic distribution and Inter-Sample variation of Non-CpG methylation across human cell types. PLoS Genetics 2011; 7:e1002389