PREIMPLANTATION GENETIC TESTING AND BIOPSY TECHNIQUES
Paula Brígido Llinares, MSc1,2; Roberto de la Fuente Pita, Ph.D1; Javier del Río Riego1, MSc; and Sara Sanz Juste, MSc1,3.
1 Embryologist Media, online non-profit website about Reproduction Biology, Spain.
2 Bioarray Edificio Quórum III, Av de la Universidad s/n, 03202, Elche, Alicante, Spain.
3 Fertility Specialists Medical Group, 8010 Frost St suite p, San Diego, California.
Contact: Javier del Río Riego (delrioriegojavier@gmail.com); Sara Sanz Juste (sara.sanz.juste@gmail.com).
Currently, Preimplantation genetic testing provides an alternative to prenatal diagnosis to detect specific genetic conditions. This approach usually is performed in advanced maternal age or to avoid inherited genetic conditions. Depending on the case and the center in which the biopsy is performed different types of biopsies can be done. Finally, considering the reason of this study, a study of aneuploidies, structural chromosomal rearrangements or monogenic diseases will be performed, and the presumed unaffected embryo(s) will be transferred or cryopreserved for later use.
Introduction
Preimplantation genetic testing (PGT) provides an alternative to prenatal diagnosis to detect specific genetic conditions and allowing the achievement of a pregnancy and birth of an unaffected child of the studied mutations (1). This test requires the analyses of the embryos generated by assisted reproductive technology (ART), by means of accurate and sensitive methodologies such as embryo biopsy, genetics and of course, background on preimplantation diagnosis and counseling from experts (2).
Clinical application of PGT dates back to the late 60’s, when blastocysts of research animals could be sexed (3), this happened ten years before Louise Brown, the first baby born by in vitro fertilization (IVF) in 1978. At the beginning of the 90’s, early human embryos were sexed before implantation and the first genetic analyses were performed to avoid children inheriting Mendelian diseases. By the end of the century, the genetic methodologies, now considered to be basic, were routinely used for PGT which was applied as a normal procedure for trying to achieve unaffected live births (4).
In the present review, we aim to give an account of the importance of PGT and the current view of the main clinical approaches for its application.
When is PGT indicated?
There is a multitude of indications for PGT which emerge from different motivations. Firstly, the patient may have suffered from a number of terminations due to the embryo having inherited the genetic condition. It could also be motivated by the parents already have a child with a severe genetic disease. In this case, they may be looking to avoid repeating this scenario or may wish to have a second baby genetically selected to save the life of their sick child (2).
Secondly, if the parents are carriers of any genetic disease, either an autosomal-dominant disorder like Huntington’s disease or an autosomal-recessive one like cystic fibrosis, they are at reproductive risk because the resulting embryo may be affected. Beyond these biological concerns, there can be ethical or religious indications, for example, certain families might have serious concerns about going on for an abortion of an affected embryo. In such cases, application of PGT may circumvent this kind of ethical conflict (2).
Finally, another indication for PGT is advanced maternal age. It is associated with an increase in oocyte abnormalities which leads to higher incidence of embryo aneuploidy. This has been implicated in low embryo implantation and higher miscarriage rates (5). However, there are still some limitations to the procedure in this patient group, as the number of embryos obtained from such couples is not always sufficient to select even a single unaffected embryo (1).
Applying PGT
Broadly speaking, steps for PGT are as follows (2) and a summary can be seen in Fig.1:
- Standard ART treatment to collect and fertilize oocytes.
- Embryo culture up to the desired point of development.
- Biopsy and embryo vitrification (if applicable).
- Genetic testing to confirm the presence or absence of the genetic condition.
- The presumed unaffected embryo(s) is/are warmed and transferred into the uterus.
- Spare unaffected embryos for the studied mutations can remain cryopreserved for later use, whereas affected embryos are discarded. This means that they can perish, although specific procedures may depend on local regulations.
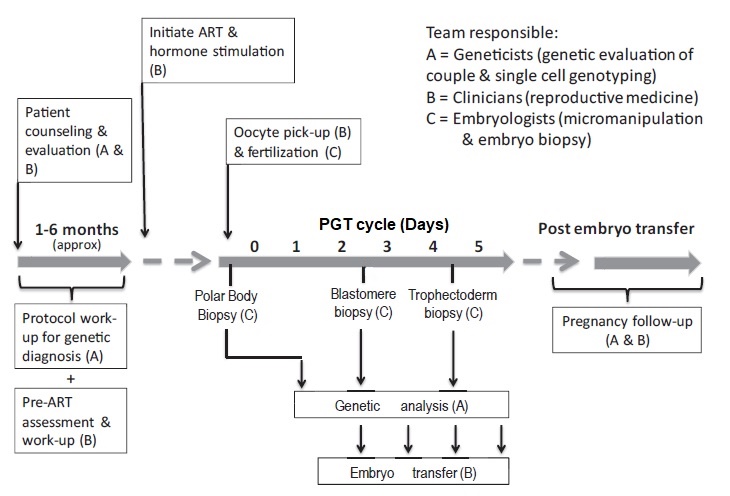
Fig. 1 Timeline of a typical PGT cycle (6).
Currently PGT terms
Over the last years of PGT application, the technology has been significantly improved, with distinct technologies being developed for specific applications: including PGT-A for aneuploidies, PGT-SR for structural rearrangements, and PGT-M for monogenic diseases and preimplantation testing for non-genetic conditions, such as HLA typing (1). Previously, these were termed preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS), whereby PGS was developed to confirm the ploidy status of the embryo, and PGD included genetic tests which determine whether an embryo is affected by a specific genetic condition (6).
PGT-A
In the past, PGS was indicated for aneuploidy detection. Now with the new terms, PGT-A replaces this objective. Over time, the improvement of molecular techniques, such as next-generation sequencing or array, has improved the diagnosis of these diseases. Therefore, the application of next-generation technologies represents great progress in ART results, particularly for couples of advanced reproductive age (1). It has been shown that transfer of one euploid blastocyst increase the live-birth rate after PGT-A cycles (8).
PGT-SR
PGT is also used in cases of structural chromosomal rearrangements. This is very useful in those carriers of balanced translocations since they have a high probability of having an affected child. In these cases, the results vary depending on the origin and type of translocation, with poorer clinical results in reciprocal translocations than Robertsonian translocations (7).
PGT-M
Monogenic diseases such as cystic fibrosis and X-linked conditions represent a further application for PGT. However, in some cases, erroneous diagnoses were detected (1). These were due to allele-specific amplification failure (allele drop-out or ADO), which implies that this type of analysis requires the development of a specific protocol for reliable detection (7).
Types of approaches for PGT in the laboratory
Available data suggest that the majority of first trimester miscarriages are a consequence of some sort of aneuploidies (7, 9). Thus, the main approach developed for PGT was the fluorescence in situ hybridization (FISH) for such chromosomes. Currently, new molecular techniques have substituted this molecular technique since these have a greater reliability than FISH (1). Current technical methodologies for PGT mainly lie in one of the followings (10,11,12):
Single-cell array comparative genomic hybridization (a-CGH)
Based on DNA templates onto microarrays, a-CGH allows for the simultaneous screening of all 24 chromosomes from embryo cells, either looking for an aneuploidy or for DNA copy-number aberrations (10,11,12).
Single Nucleotide Polymorphism Microarray (SNP)
Single nucleotide polymorphism detects pairs of single nucleotides in genomic DNA, approximately 250 common structural aberrations and uniparental disomy. However, SNP arrays for PGT have been found to be time-consuming, costly, and complex (10,11,12).
Next Generation Sequencing (NGS)
NGS is a high throughput technique that can evaluate samples for multiple indications on the same chip (ideal for PGT-A and suitable for low DNA input). It not only screens for aneuploidies but can also detect both nuclear single-gene disorders and mitochondrial abnormalities (10,11,12).
Biopsy options
Currently, different types of biopsies can be done, depending on the case and the center in which the biopsy is performed. Typical biopsies for PGT are described below:
Polar body biopsy
Genetic analysis of the polar body extruded from the oocyte gives an accurate chromosomal evaluation of the oocyte. In monogenic diseases cases or chromosomal alterations diagnosis, the results of both the first and second polar bodies are required (6), which can be done either sequentially or simultaneously (Fig. 2). Significant advantages of PGT were achieved by the performance of polar body biopsy (13).
Unfortunately, this approach gives no information on the paternal contribution to the embryo, so it is unable to specify whether the participating sperm presents any genetic abnormality (13). Also, the first polar body may degenerate before the second polar body is biopsied, preventing the achievement of a conclusive diagnosis (6). Considering these limitations, why is this technique performed? This technique is commonly performed due to ethical concerns or restrictions regarding embryo biopsy in the country the analyses are being carried out (13). But also, it can be due to scientific reasons, since it has been said that it is less invasive than blastomere or trophectoderm biopsy and avoids any potential harm to the embryo (14).
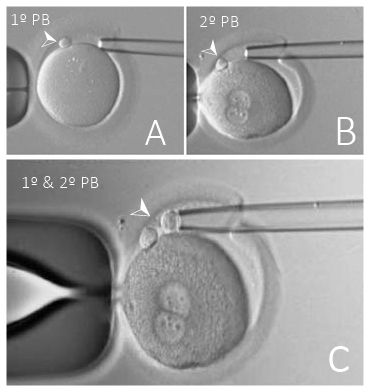
Fig. 2 Polar body biopsies (PB). (A) First polar body removal, (B) Second polar body removal,
(C) First and second polar body removal. Modified from (15).
Blastocentesis
A few pieces of research about the partial suction of the blastocoel fluid (BF) for genomic DNA extraction have proposed blastocentesis as a new method for PGT (16,17,1). BF contains cell-free DNA, mostly as a result of programmed cell death or apoptosis during the normal regulation of the developing embryo (18). However, while aspiration of BF is less invasive than trophectoderm biopsy, it is not completely non-invasive, and its accuracy has not yet been demonstrated (1), Fig. 3.
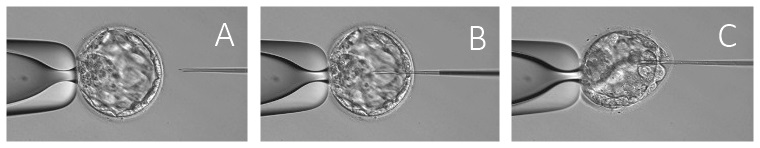
Fig. 3 Partial suction of the BF for genomic DNA extraction. (A) The ICSI needle is (B) inserted through the zona pellucida, followed by
(C) aspiration to allow the blastocoel cavity to collapse around the needle while retracting the needle. Modified from (19).
Cell-free DNA in the medium
Finally, Hammond et al. had suggested that the culture media contains DNA molecules secreted by the embryo DNA originating from oocyte/embryo-associated cells and organelles, such as granulosa cells or polar bodies (20), Fig. 4. This has been suggested to be caused by a repair mechanism used by embryos to avoid aneuploidies (20). This could mean that a biopsy is not necessary for a chromosomal diagnosis since the media may have the sample to analyse. However, the percentages of informative samples showed in the literature vary widely, being reported values at 3.5% (21), 27% (22) and 85.7% (23). Also, there is evidence that the difference in the amount of DNA into the cultures of aneuploid and euploid embryos is limited (24). These discrepancies, with the chance of maternal DNA contamination and the mosaicism that exists in the embryo trophectoderm biopsy (20) are some of the issues that are necessary to overcome before using cell-free DNA as a representative source for non-invasive PGT.
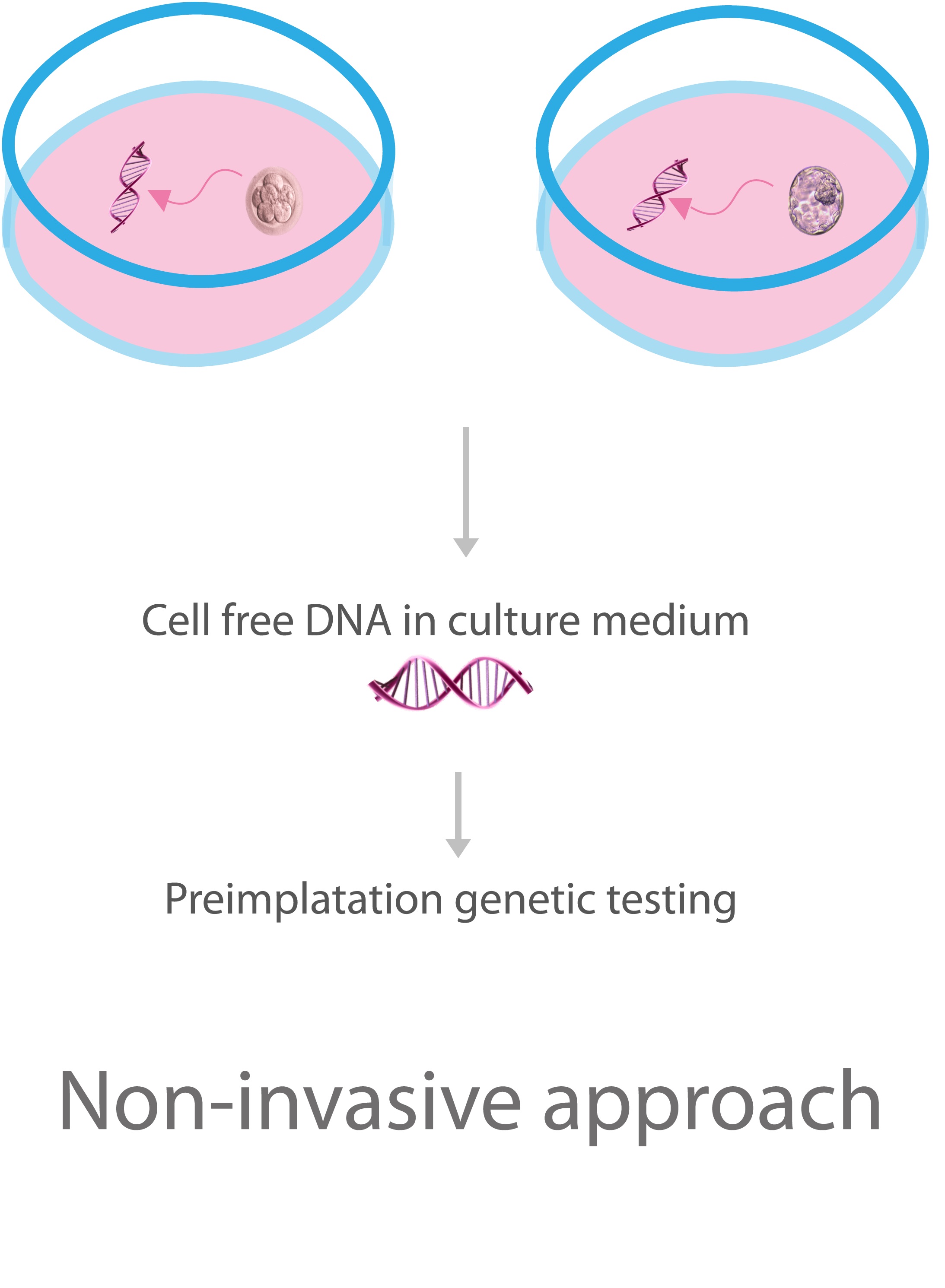
Fig. 4 PGT with cell-free DNA in the medium. Adapted from (25).
Day 3. Cleavage stage biopsy
Embryos are biopsied at day 3 since the individual cells can be differentiated (26). Moreover, the cells are still totipotent and are usually not yet adhering to one another (1). This technique entails aspiration of one or two blastomeres to obtain the embryonic genetic material for PGT (24), as can be seen in the Fig. 5. To perform the biopsy, it usually requires a brief exposure of the embryos to Ca2+/Mg2+-free media to reduce adherence between cells in order to facilitate the technique (6). This method allows the embryos to be cultured in vitro until they reach the blastocyst stage and to fresh embryo transfer. Furthermore, blastomere biopsy is advantageous compared with polar body biopsy because both maternal and paternal genetic contributions to the embryo can be analysed (27).
On the other hand, there is some controversy regarding the use of this type of biopsy because of the amount of genetic material in a single blastomere for analysis is limited (6). Also, the cleavage biopsy is more expensive than the blastocyst biopsy since there are more embryos to study and the samples must be transported urgently to the genetic laboratory (28).
- How many cells should be removed?
The number of cells to be removed in the biopsy is still a controversial issue. Aspirating one cell reduces the cellular mass extracted but results can be skewed by mosaicism (Fig 5). Conversely, aspirating two cells can reduce the risk of mosaicism, but removing such cellular mass could have consequences on the implantation rate (28).Reported data have shown a dramatic reduction of 39% in the implantation rate after a cleavage-stage biopsy (29). The authors related it to the proportion of the embryo total cellular removed. Whereas around 5 cells pulled out of the embryo in the trophectoderm biopsy represent 2-3% of the total cell content (expanded blastocyst has 200-220 cells approximately), extraction of a single cell from an eight-cell embryo supposes 13% of the total content (29).
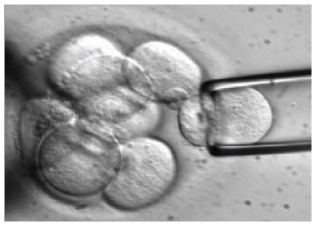
Fig. 5 Cleavage-stage biopsy. Modified from (10).
- What does the literature say?
Cleavage-stage biopsy produces different opinions among the experts because of the presence of mosaicism and the possibility of self-correction of aneuploidies from cleavage to the blastocyst stage (30). On the contrary, studies using a-CGH technology to analyse genetic abnormalities in day-3 blastomeres and confirming it in trophectoderm biopsy showed concordance between day 3 diagnosis and day 5 re-analysis. Also, Treff and coauthors showed more reliable results for SNP-microarray (96% vs. 83%) and a lower mosaicism degree (31%) for SNP-microarray samples in a study comparing array technology versus FISH technique (31). These data would support the suggestion of some authors, who proposed that the incidence of mosaicism may have been overestimated in previous studies due to the technical inconsistency of the FISH technique (30, 31, 32). At present, this matter remains controversial.
Day 5. Trophectoderm biopsy
The blastocyst stage is currently thought to be the optimal time to perform a biopsy (33). The combination of improved blastocyst culture, TE biopsy, refined cryopreservation techniques, and molecular assays, such as array comparative genomic hybridization that allows for 24-chromosome screening, have led to a renaissance of PGT in this stage (33, 34).
TE biopsy will not detect every incidence in which the embryo has risk of aneuploidy, but it will detect mosaicism more reliably than the cleavage-stage biopsy (33, 34). When PGT-M is performed in the stage of cleavage, it exists a high risk of failure due to ADO. Consequently, the number of healthy embryos that could be transferred is underestimated (28).
By performing the biopsy in blastocyst stage, there is an increase in the amount of starting DNA template, which should enhance the sensitivity and reliability of genetic diagnosis (Fig. 6). Therefore, it leads to improved PGT outcome for patients (28). Also, it is of a great importance to choose a solid method for detecting potential ADO. There is evidence that simultaneous amplifications and the additional linked marker may reduce the diagnosis errors rate by half (1, 35).
- How many cells should be removed?
Research to determine the appropriate number of biopsied TE cells in blastocyst biopsies is limited. The exact number of biopsied TE cells is hard to count visually because cells are small and usually remain as a clump. In most studies using a-CGH or SNP for genetic testing, biopsied TE cells were used for genome amplification and their number was impossible to know. Moreover, some studies showed that removing 1-5 cells may be the appropriate biopsied TE cell number to maintain the implantation potential (27, 36, 37).
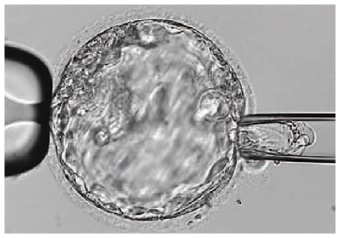
Fig. 6 Blastocyst-stage biopsy.
Modified from (10).
- Can biopsies affect blastocyst development and its implantation?
Whereas it remains possible that biopsy of cleavage-stage embryos can critically arrest further development through reduction of cell mass, the low miscarriage rates and high term birth rates suggest that this is not the case for TE biopsy. It can be speculated that the damage to blastocyst development potential caused by TE biopsy would be less for blastocysts with a greater number of TE cells (34, 36).Some experts assured that TE biopsy at the blastocyst stage had no meaningful impact on the developmental competence of the embryo as measured by implantation and delivery rates (27). When combined with TE biopsy and blastocyst vitrification, SNP microarray has resulted in high implantation and low miscarriage rates for some IVF patients (27, 29, 38).
- Are there any limitations?
Owing to the limitations of genetic analysis, most of the biopsied blastocysts need to be cryopreserved by vitrification, and blastocysts with normal results would be transferred in the next frozen cycle. In addition, biopsy of numerous cells from blastocysts with grade B or C may cause damage to the embryo, leading to either its arrest or implantation failure (27, 36).Also, the personal experience of different embryologists is an influencing factor in this technique. The number of biopsied cells in the blastocyst biopsy is hard to quantify and largely dependent on the experience of embryologist (36).
Conclusion
The availability of new approaches provides the opportunity to offer patients reduced risk of transferring embryos with genetic issues. The cleavage-stage biopsy allows for fresh embryo transfer after genetic diagnosis. However, there are reports of high levels of mosaicism when the biopsy is performed on day 3. Trophectoderm biopsy, in turn, provides a greater amount of material for an effective and more reliable diagnosis in embryos compared to those on cleavage stage. The drawback for this option is the usual need for cryopreservation and transfer in a different cycle.
The change of biopsy procedures, together with the application of vitrification techniques, has contributed greatly to the improvement of the reproductive outcome of the PGT. Further improvement of biopsy and freezing procedures will be achieved to minimize or totally avoid embryo damage, while future research in this field should make possible to develop less invasive PGT approaches. Finally, it is important to take in consideration not only the technical aspects if not also the wishes of the patient.
Referencias
1. Kuliev A, Rechitsky S. Preimplantation genetic testing: current challenges and future prospects. Expert Rev Mol Diagn. 2017;17(12):1071-1088.
2. Braude P, Pickering S, Flinter F, Ogilvie C. Preimplantation genetic diagnosis. Nat Rev Genet. 2002; 3(12):941-53.
3. Gardner RL, Edwards RG. Control of the sex ratio at full term in the rabbit by transferring sexed blastocysts. Nature. 1968; 218(5139):346-9.
4. Preimplantation Genetic Diagnosis International Society. http://www.pgdis.org/history.html.
5. Rubio C, Bellver J, Rodrigo L, Castillón G, Guillén A, Vidal C et al. In vitro fertilization with preimplantation genetic diagnosis for aneuploidies in advanced maternal age: a randomized, controlled study. Fertil Steril. 2017;107(5):1122-1129.
6. Traeger-Synodinos J. Pre-implantation genetic diagnosis. Best Pract Res Clin Obstet Gynaecol. 2017;39:74-88.
7. Brezina P, Brezina D, Kearns W. Preimplantation genetic testing. BMJ. 2012; 345:e5908.
8. Ubaldi F, Cimadomo D, Capalbo A, Vaiarelli A, Buffo L, Trabucco E et al. Preimplantation genetic diagnosis for aneuploidy testing in women older than 44 years: a multicenter experience. Fertil Steril. 2017;107(5):1173-1180.
9. Kearns W, Pen R, Graham J, Han T, Carter J, Moyer M et al. Preimplantation genetic diagnosis and screening. Semin Reprod Med. 2005; 23(4):336-47.
10. Sullivan-Pyke C, Dokras A. Preimplantation Genetic Screening and Preimplantation Genetic Diagnosis. Obstet Gynecol Clin N Am. 2018;45(1):113-125.
11. Li G, Niu W, Jin H, Xu J, Song W, Guo Y et al. Importance of embryo aneuploidy screening in preimplantation genetic diagnosis for monogenic diseases using the karyomap gene chip. Sci Rep. 2018;8(1).
12. Capalbo A, Romanelli V, Cimadomo D, Girardi L, Stoppa M, Dovere L et al. Implementing PGD/PGD-A in IVF clinics: considerations for the best laboratory approach and management. J Assist Reprod Genet. 2016; 33(10):1279-286.
13. Brezina PR, Ke, RW, Kutteh WH. Preimplantation Genetic Screening: A Practical Guide. Clin Med Insights Reprod Health. 2013;7:37-42.
14. Schenk M, Groselj-Strele A, Eberhard K, Feldmeier E, Kastelic D, Cerk S et al. Impact of polar body biopsy on embryo morphokinetics-back to the roots in preimplantation genetic testing?. J Assist Reprod Genet. 2018.
15. Available from: https://www.glowm.com/resources/glowm/cd/pages/v5/ch109/framesets/001f.html [Cited 11 June 2018].
16. Chen S, Lee T, Lien Y, Tsai Y, Chang L, Yang Y. Microsuction of blastocoelic fluid before vitrification increased survival and pregnancy of mouse expanded blastocysts, but pretreatment with the cytoeskeletal stabilizer did not increase blastocyst survival. Fertil Steril. 2005; 84:1156-62.
17. Gianaroli L, Magli MC, Stanghellini I, Crippa A, Crivello AM, Pescatori ES et al. DNA integrity is maintained after freeze-drying of human spermatozoa. Fertil Steril. 2012; 97(5):1067-73
18. Palini S, Galluzzi L, De Stefani S, Bianchi M, Wells D, Magnani M et al. Genomic DNA in human blastocoele fluid. Reproductive BioMedicine Online. 2013; 26:603-10.
19. Tobler K, Zhao Y, Ross R, Benner A, Xu X, Du L et al. Blastocoel fluid from differentiated blastocysts harbors embryonic genomic material capable of a whole-genome deoxyribonucleic acid amplification and comprehensive chromosome microarray analysis. Fertil Steril. 2015;104(2):418-425.
20. Hammond ER, McGillivray BC, Wicker SM, Peek JC, Shelling AN, Stone P, et al. Characterizing nuclear and mitochondrial DNA in spent embryo culture media: genetic contamination identified. Fertil Steril. 2017;107(1):220-228.e5.
21. Shamonki MI, Jin H, Haimowitz Z, Liu L. Proof of concept: preimplantation genetic screening without embryo biopsy through analysis of cell-free DNA in spent embryo culture media. Fertil Steril. 2016;106(6):1312-8.
22. Feichtinger M, Vaccari E, Carli L, Wallner E, Mädel U, Figl K, et al. Non-invasive preimplantation genetic screening using array comparative genomic hybridization on spent culture media: a proof-of-concept pilot study. Reprod Biomed Online. 2017;34(6):583-9.
23. Xu J, Fang R, Chen L, Chen D, Xiao J-P, Yang W, et al. Noninvasive chromosome screening of human embryos by genome sequencing of embryo culture medium for in vitro fertilization. Proc Natl Acad Sci U S A. 18 de 2016;113(42):11907-12.
24. Vera-Rodriguez M, Diez-Juan A, Jimenez-Almazan J, Martinez S, Navarro R, Peinado V, et al. Origin and composition of cell-free DNA in spent medium from human embryo culture during preimplantation development. Hum Reprod Oxf Engl. 2018;33(4):745-56.
25. Assou S, Ait-Ahmed O, El Messaoudi S, Thierry AR, Hamamah S. Non-invasive pre-implantation genetic diagnosis of X-linked disorders. Med Hypotheses. 2014; 83:506–508.
26. Harper JC, Boelaert K, Geraedts J, Harton G, Kearns WG, Moutou C et al. ESHRE PGD Consortium data collection V: cycles from January to December 2002 with pregnancy follow-up to October 2003. Hum Reprod. 2006; 21:3–21.
27. Jing S, Luo K, He H, Lu C, Zhang S, Tan Y et al. Obstetric and neonatal outcomes in blastocyst-stage biopsy with frozen embryo transfer and cleavage-stage biopsy with fresh embryo transfer after preimplantation genetic diagnosis/screening. Fertil Steril. 2016; 106(1):105-112.e4.
28. Kokkali G, Traeger-Synodinos J, Vrettou C, Stavrou D, Jones G, Cram D et al. Blastocyst biopsy versus cleavage stage biopsy and blastocyst transfer for preimplantation genetic diagnosis of β-thalassaemia: a pilot study. Hum Reprod. 2007;22:1443–9.
29. Scott R, Upham K, Forman E, Zhao T, Treff N. Cleavage-stage biopsy significantly impairs human embryonic implantation potential while blastocyst biopsy does not: a randomized and paired clinical trial. Fertil Steril. 2013;100(3):624-630.
30. Rubio C, Rodrigo L, Mir P, Mateu E, Peinado V, Milán M et al. Use of array comparative genomic hybridization (array-CGH) for embryo assessment: clinical results. Fertil Steril. 2013; 99(4):1044-8.
31. Treff NR, Levy B, Su J, Northrop LE, Tao X, Scott Jr RT. SNP microarray-based 24 chromosome aneuploidy screening is significantly more consistent than FISH. Mol Hum Reprod. 2010;16(8):583–9.
32. Mir P, Mateu E, Mercader A, Herrer R, Rodrigo L, Vera M et al. Confirmation rates of array-CGH in day-3 embryo and blastocyst biopsies for preimplantation genetic screening. J Assist Reprod Genet. 2016;33(1):59-66.
33. Grifo J, Adler A, Lee H, Morin S, Smith M, Lu L et al. Deliveries from trophectoderm biopsied, fresh and vitrified blastocysts derived from polar body biopsied, vitrified oocytes. Reprod Biomed Online. 2015; 31(2):210-216.
34. McArthur S, Leigh D, Marshall J, de Boer K, Jansen R. Pregnancies and live births after trophectoderm biopsy and preimplantation genetic testing of human blastocysts. Fertil Steril. 2005; 84(6):1628-1636.
35. ESHRE Special Interest Group of Embryology, Alpha Scientists in Reproductive Medicine. The Vienna consensus: report of an expert meeting on the development of art laboratory performance indicators. Hum Reprod Open. 2017;(2).
36. Zhang S, Luo K, Cheng D, Tan Y, Lu C, He H et al. Number of biopsied trophectoderm cells is likely to affect the implantation potential of blastocysts with poor trophectoderm quality. Fertil Steril. 2016; 105(5):1222-1227.e4.
37. Christodoulou C, Dheedene A, Heindryckx B, van Nieuwerburgh F, Deforce D, De Sutter P et al. Preimplantation genetic diagnosis for chromosomal rearrangements with the use of array comparative genomic hybridization at the blastocyst stage. Fertil Steril. 2017; 107(1):212-219.e3.
38. Schoolcraft W, Fragouli E, Stevens J, Munne S, Katz-Jaffe M, Wells D. Clinical application of comprehensive chromosomal screening at the blastocyst stage. Fertil Steril. 2010; 94(5):1700-1706.